Phase III Randomized Trial of Single vs. Tandem Myeloablative Consolidation Therapy for High-Risk Neuroblastoma
A Groupwide Phase III Study
THIS PROTOCOL IS FOR RESEARCH PURPOSES ONLY, AND SHOULD NOT BE COPIED, REDISTRIBUTED OR USED FOR ANY OTHER PURPOSE. MEDICAL AND SCIENTIFIC INFORMATION CONTAINED WITHIN THIS PROTOCOL IS NOT INCLUDED TO AUTHORIZE OR FACILITATE THE PRACTICE OF MEDICINE BY ANY PERSON OR ENTITY. RESEARCH means a systematic investigation, including research development, testing and evaluation, DESIGNED TO DEVELOP OR CONTRIBUTE TO GENERALIZABLE KNOWLEDGE. THIS PROTOCOL IS THE RESEARCH PLAN DEVELOPED BY THE CHILDREN'S ONCOLOGY GROUP TO INVESTIGATE A PARTICULAR STUDY QUESTION OR SET OF STUDY QUESTIONS AND SHOULD NOT BE USED TO DIRECT THE PRACTICE OF MEDICINE BY ANY PERSON OR TO PROVIDE INDIVIDUALIZED MEDICAL CARE, TREATMENT, OR ADVICE TO ANY PATIENT OR STUDY SUBJECT. THE PROCEDURES IN THIS PROTOCOL ARE INTENDED ONLY FOR USE BY CLINICAL ONCOLOGISTS IN CAREFULLY STRUCTURED SETTINGS, AND MAY NOT PROVE TO BE MORE EFFECTIVE THAN STANDARD TREATMENT. ANY PERSON WHO REQUIRES MEDICAL CARE IS URGED TO CONSULT WITH HIS OR HER PERSONAL PHYSICIAN OR TREATING PHYSICIAN OR VISIT THE NEAREST LOCAL HOSPITAL OR HEALTHCARE INSTITUTION.
SEE SECTIONS 14 AND 15 FOR SPECIMEN SHIPPING ADDRESSES
Abstract
The Children's Oncology Group has received a Certificate of Confidentiality from the federal government, which will help us protect the privacy of our research subjects. The Certificate protects against the involuntary release of information about your subjects collected during the course of our covered studies. The researchers involved in the studies cannot be forced to disclose the identity or any information collected in the study in any legal proceedings at the federal, state, or local level, regardless of whether they are criminal, administrative, or legislative proceedings. However, the subject or the researcher may choose to voluntarily disclose the protected information under certain circumstances. For example, if the subject or his/her guardian requests the release of information in writing, the Certificate does not protect against that voluntary disclosure. Furthermore, federal agencies may review our records under limited circumstances, such as a DHHS request for information for an audit or program evaluation or an FDA request under the Food, Drug and Cosmetics Act. The Certificate of Confidentiality will not protect against mandatory disclosure by the researchers of information on suspected child abuse, reportable communicable diseases, and/or possible threat of harm to self or others.
Abstract
ANBL0532 is a Phase III trial to study the use of intensified consolidation (Primary Aim 1), the use of newer agents during induction (Primary Aim 2), and the use of high dose local radiation (Primary Aim 3).
The primary goal of ANBL0532 is to test whether further intensification of myeloablative therapy will improve cure rate for high risk neuroblastoma.
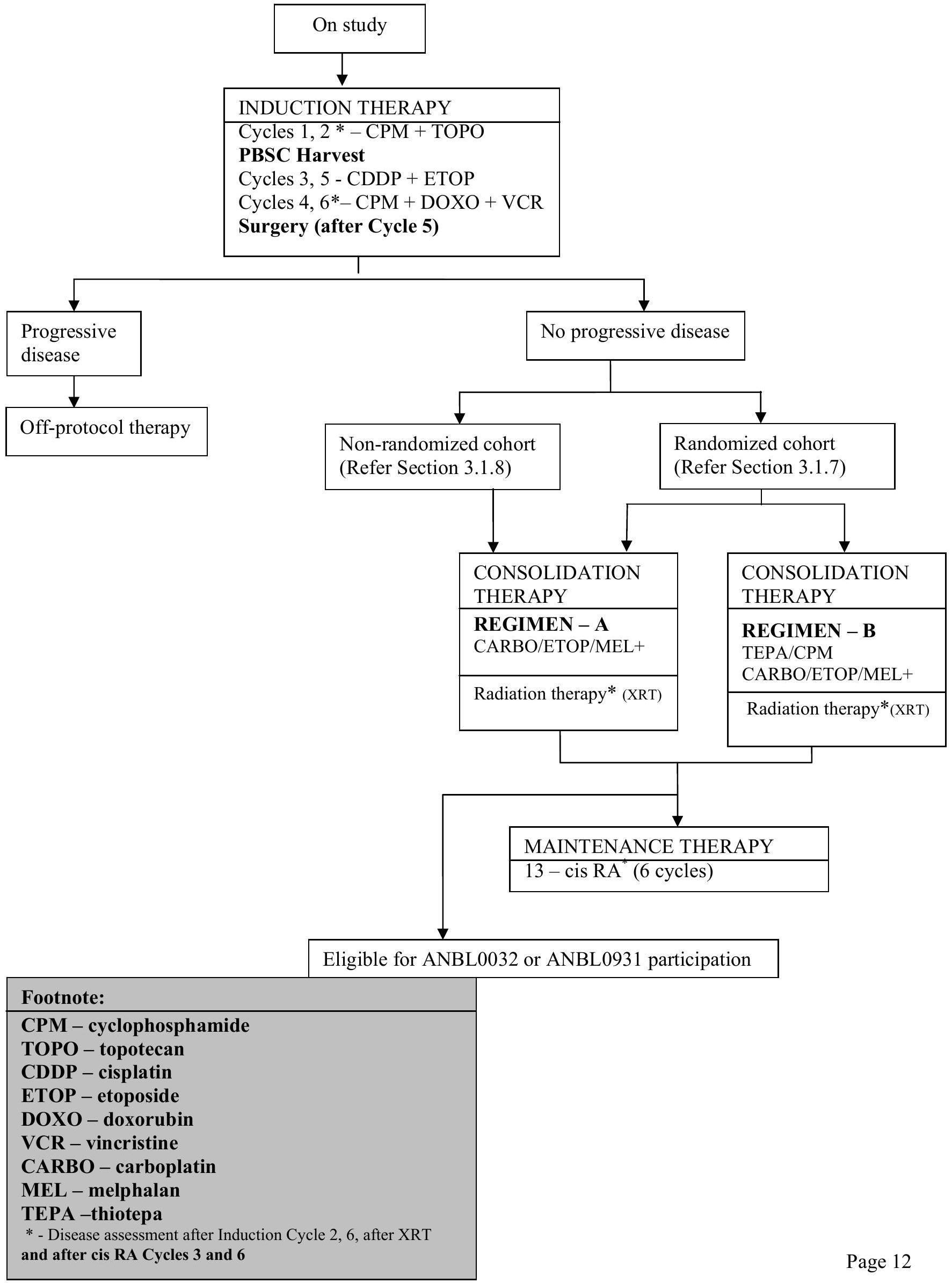
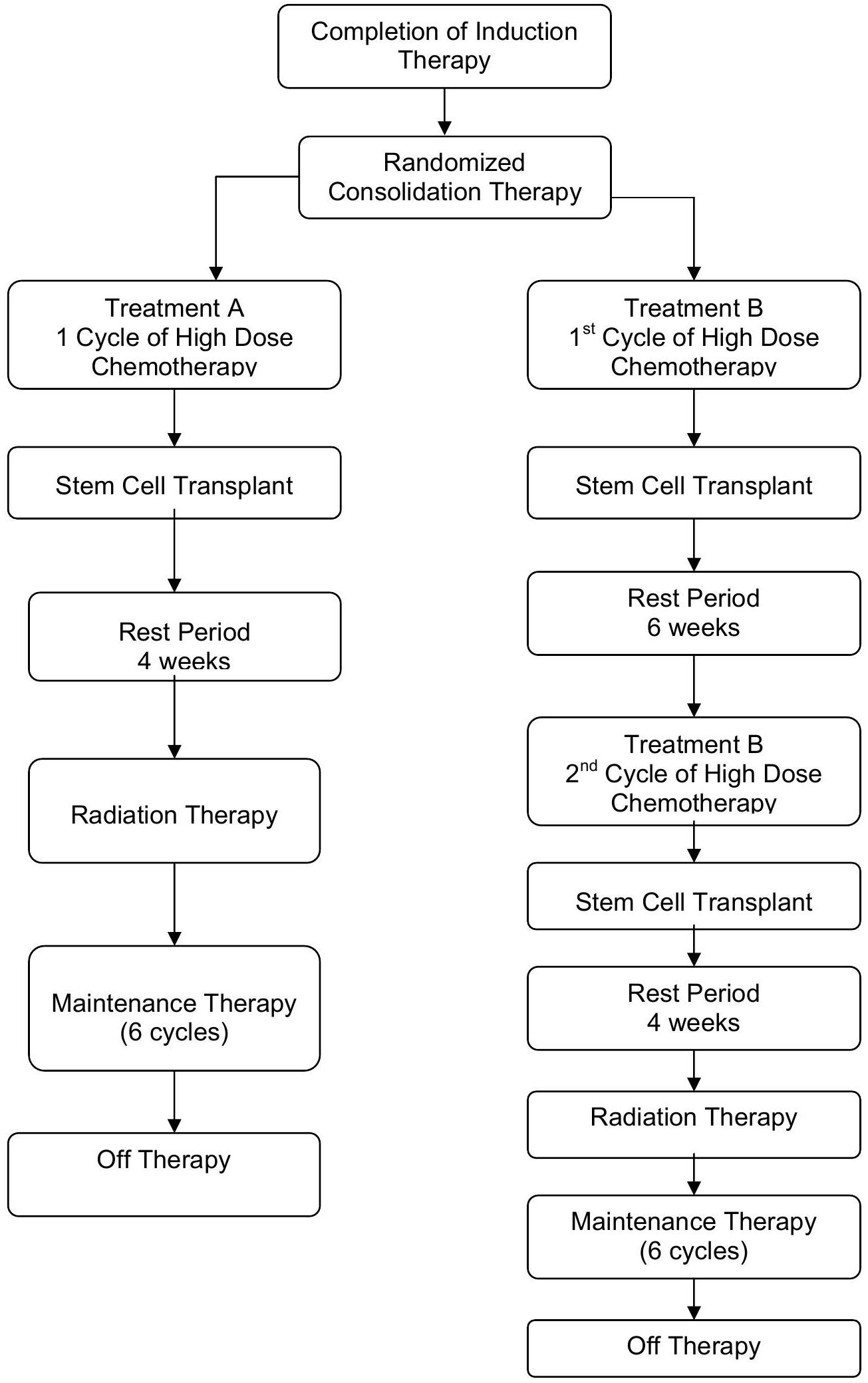
Myeloblative consolidation therapy is given at the completion of induction therapy and is categorized into Regimen A and Regimen B. Regimen A consists of one myeloablative consolidation with a carboplatin etoposide - melphalan (CEM) preparative regimen. For Regimen B, patients will receive two myeloablative consolidations: Thiotepa - cyclophosphamide (TC) preparative regimen followed by CEM preparative regimen.
Most patients will be randomized into either Regimen A or Regimen B. Randomization will be stratified by stage, MYCN status and response to induction therapy. Patients 365 to 547 days of age ( 12 - 18 months) with Stage 4, MYCN nonamplified tumor with unfavorable histopathology or diploid DNA content or with indeterminant histology or ploidy and patients who are greater than 547 days of age with Stage 3, MYCN nonamplified tumor AND unfavorable histopathology or indeterminant histology will be nonrandomly assigned to single myeloablative transplant (Regimen A).
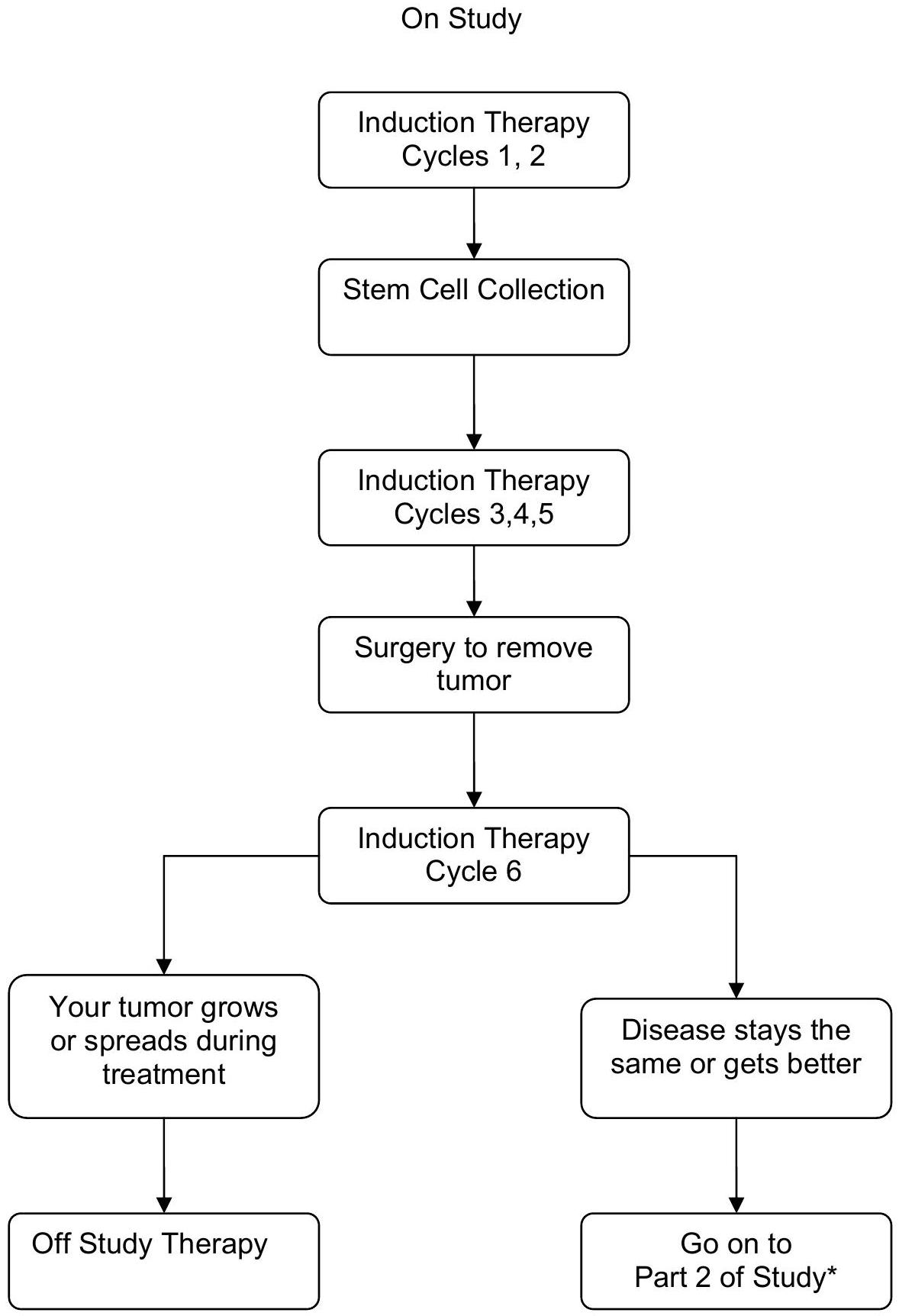
ANBL0532 will also assess the efficacy of a dose intensive topotecan containing induction regimen, substituting 2 cycles of dose intensive cyclophosphamide and topotecan for the initial 2 cycles of induction utilized on the prior COG A3973 protocol. Patients will receive 6 cycles of induction chemotherapy as outlined in the schema below. Patients will undergo peripheral blood stem cell (PBSC) harvest after 2 cycles with no ex vivo manipulation prior to cryopreservation. PBSC collection will be performed prior to randomization. The goal of apheresis will be collection of divided into 3 aliquots. A minimum collection of , divided into 2 aliquots is required for each patient while an additional aliquot of is strongly recommended to be stored as backup for delayed engraftment or for future use. Tumor response will be assessed after 2 cycles of induction chemotherapy. Patients will undergo surgical resection after Cycle 5 (or 6 if necessary). Tumor response will be assessed after completion of surgical resection and 6 cycles of chemotherapy (end-induction). Patients with progressive disease (PD) at end of induction evaluation will be taken off protocol therapy, all other patients will continue on therapy.
ANBL0532 will also test whether additional radiation therapy delivered to gross residual disease improves local control for those patients with less than a gross total resection. After completion of transplant and recovery from acute toxicities, patients will undergo radiation to primary site of disease
and to MIBG-avid sites seen at pre-transplant (end-induction) evaluation. Patients who achieve end of induction primary site complete response (CR) will receive 21.6 Gy external beam radiation therapy (EBRT) to the primary site (standard dose) while patients achieving ( residual soft tissue) at the primary site will receive an additional boost of 14.4 Gy for a total dose of 36 Gy EBRT delivered to gross residual primary site disease. Pre-surgical and end of induction CT or MRI scans and operative reports will be centrally reviewed in real time for all patients assessed by the treating institution to have a complete response at the primary site at the end of induction. Tumor response will be assessed after completion of radiation therapy.
Patients will be encouraged to enroll onto ANBL0032 or ANBL0931for Maintenance biologic therapy. Alternatively, post-transplant Maintenance therapy with cis-RA daily for 14 days every 28 days repeated for 6 months will be administered.
Pharmacogenomics studies will be performed to evaluate whether host genetic polymorphisms in cyclophosphamide metabolizing enzymes are associated with cyclophosphamide/topotecan - related toxicity and tumor response. Cis-RA pharmacokinetics and pharmacogenomic studies will be performed to evaluate whether the variability of cis-RA continuous steady state plasma levels and/or genetic variations in retinoic acid metabolic enzymes correlate with event-free survival or systemic toxicities in high-risk neuroblastoma patients. Total topotecan levels will be assessed to formulate a population-based dosing model for topotecan in children with high-risk neuroblastoma.
Post-operative complications will be studied prospectively in patients with high risk neuroblastoma to better characterize the incidence and timing of occurrence. Specifically, the occurrence of bowel obstruction, renal atrophy, chylous leak and chronic diarrhea will be evaluated through 12 months from completion of therapy. Rates of these complications will be correlated with the surgical and radiotherapeutic efforts in obtaining local control as they may impact on future management decisions. The neurologic and orthopedic outcome for patients with intraspinal extension of primary tumors (referred to as paraspinal tumors) will be studied prospectively on this protocol, to better characterize the multiple aspects of disability that these patients may be at risk for. It is the intent of this study that patients with clinical signs of spinal cord compression will be treated with primary chemotherapy; however, multidisciplinary discussion among oncology, neurosurgery, and radiation oncology is strongly encouraged at the time of diagnosis.
Clinical outcome data for patients treated on this study will be used in conjunction with ongoing biologic studies on ANBL00B1 to identify new biological surrogate markers for disease relapse and/or disease progression. In addition, immunologic assays will be employed to assess functional cellular immunity against neuroblastoma at diagnosis and after myeloablative therapy and to enumerate T-cell lymphocyte recovery following single versus tandem myeloablative therapy.
Children's Oncology Group
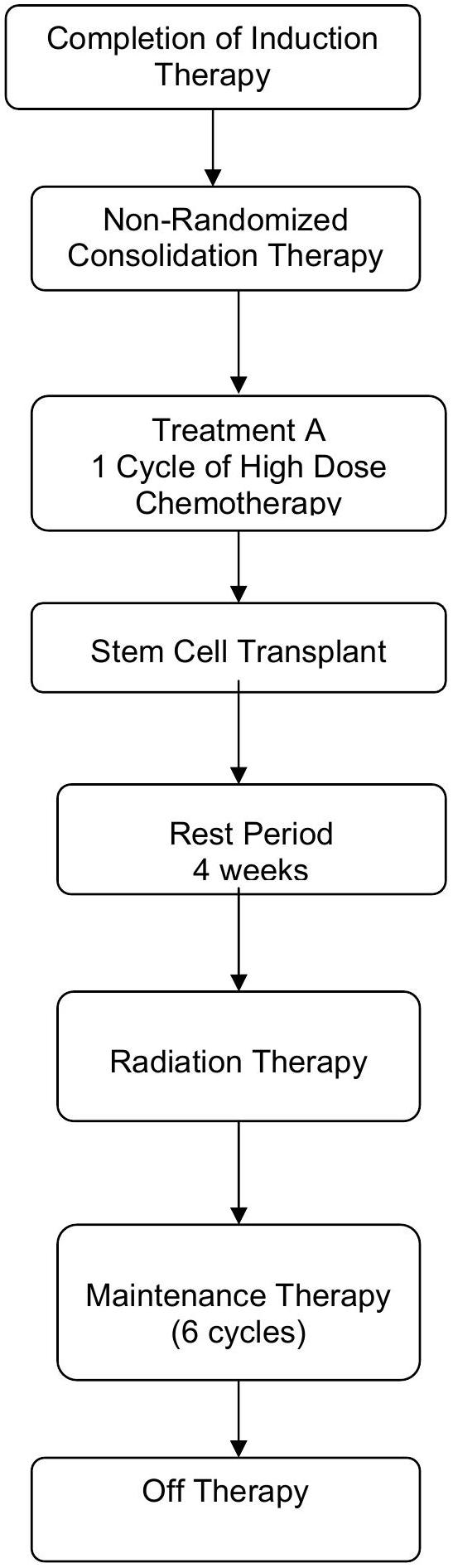
EXPERIMENTAL DESIGN SCHEMA
1.0 GOALS AND OBJECTIVES (SCIENTIFIC AIMS)
1.1 Primary Objectives
1.1.1
To improve the 3-year event free survival (EFS) rate of high-risk neuroblastoma patients through treatment with a tandem consolidation of Thiotepa/Cyclophosphamide followed by Carboplatin/Etoposide/Melphalan (CEM) as compared to single CEM consolidation.
1.1.2
To improve the rate of end-induction complete response and very good partial response, compared to historical controls, by use of a topotecan-containing induction regimen.
1.1.3
To improve the 3 -year local control rate, compared to historical controls, by increasing the local dose of radiation to the residual primary tumor for patients with less than a gross total resection.
1.2 Secondary Objectives
1.2.1
To evaluate the pharmacogenetic relationship of cyclophosphamide-metabolizing enzymes (CYP2B6, CYP2C9 and GSTA1 genotypes) with toxicity and response following dose intensive cyclophosphamide and topotecan induction chemotherapy.
1.2.2
To determine if resection completeness is predictive of a) local control rate; or b) event free survival rate in patients with high-risk neuroblastoma.
1.2.3
To prospectively describe the complications related to efforts at local control (surgery and radiation therapy) in patients with high risk neuroblastoma.
1.2.4
To describe the neurologic outcome of patients with paraspinal primary neuroblastoma tumors.
1.2.5
To determine the variability of 13-cis-retinoic-acid pharmacokinetics and relationship to pharmacogenomic parameters and determine if pharmacokinetics and/or genetic variations correlate with EFS or systemic toxicity as follows:
a) To determine the variability of 13 -cis-retinoic-acid pharmacokinetics and relationship to pharmacogenomic parameters.
b) To determine if 13-cis-retinoic-acid pharmacokinetic levels are predictive of the EFS rate or associated with systemic toxicity following 13-cis-retinoic acid.
c) To determine if pharmacogenomic variations are predictive of the EFS rate or associated with systemic toxicity following 13-cis-retinoic acid.
1.2.6
To evaluate total topotecan pharmacokinetics and correlate with patient specific data for use in an ongoing topotecan population pharmacokinetic analysis.
1.2.7
To evaluate the presence and function of T cells capable of recognizing neuroblastoma by assessing:
Children's Oncology Group
a) If T cells recognizing the neuroblastoma antigen survivin circulate at diagnosis;
b) If these T cells can be expanded using autologous antigen presenting cells (APCs);
c) If these T cells will kill neuroblastoma cells as detected in functional assays; and
d) If the presence and activity of anti-neuroblastoma immunity is decreased by stem cell transplant.
1.2.8
To characterize the recovery of T cell numbers after myeloablative consolidation and hematopoietic stem cell transplant (HSCT) and assess the impact of tandem myeloablative consolidation on T cell recovery.
1.2.9
To characterize minimal residual disease burden using RT-PCR evaluation of a panel of neuroblastomaspecific transcripts in patient bone marrow and peripheral blood following induction chemotherapy and after single versus tandem myeloablative chemotherapy and to evaluate impact on EFS.
1.2.10
To evaluate the EFS and OS rate for patients months with Stage 4, nonamplified tumor with unfavorable histopathology or diploid DNA content or with indeterminant histology or ploidy and patients who are greater than 547 days of age with Stage 3, MYCN nonamplified tumor AND unfavorable histopathology or indeterminant histology following treatment with single myeloablative transplant.
2.0 BACKGROUND
Neuroblastoma, a neoplasm of the sympathetic nervous system, is the second most extracranial malignant solid tumor of childhood. The current survival for a child year at diagnosis with Stage 4 neuroblastoma is only Novel therapies to improve initial disease response and treatment of minimal residual disease are required to improve survival for these children with high-risk neuroblastoma. Standard therapy for patients with high-risk neuroblastoma involves at least 4 components: Induction, Local Control, Consolidation and Treatment of minimal disease with biologic agents. The use of these 4 components has evolved over the last 20 years, based upon work by the POG and CCG, international cooperative groups and smaller cohort studies.
The primary aim of ANBL0532 is to improve event free survival for children with high-risk neuroblastoma. This study is a logical extension of the COG Neuroblastoma committee's strategic plan and will build upon our previous findings to study the use of intensified consolidation (Primary Aim 1), the use of a novel induction regimen (Primary Aim 2), and the use of increased dose local radiation therapy (Primary Aim 3). Specifically, a limited institution pilot study through COG (ANBL00P1) has demonstrated the feasibility and tolerable toxicity of the proposed tandem transplant consolidation regimen. An additional limited institution pilot study through COG (ANBL02P1) has demonstrated feasibility/tolerable toxicity of the dose intensive cyclophosphamide/topotecan administration proposed in the induction phase and ability to harvest PBSC following such chemotherapy. ANBL0532 will also test whether additional radiation therapy will improve local control for those patients with less than a gross total resection, a hypothesis derived directly from retrospective analysis of the prior CCG 3891 high-risk neuroblastoma protocol. Patients completing this study will be eligible for COG ANBL0032 or ANBL0931, a study of immunotherapy as treatment of minimal residual disease.
2.1 Definition of High-Risk Neuroblastoma
Patients eligible for ANBL0532 must have high-risk neuroblastoma as defined by risk criteria that include age, stage, and the biologic features of MYCN amplification, histopathology, and DNA ploidy (Appendix VII). The stratification algorithm to be used for ANBL0532 and ANBL0531 (Intermediate Risk Neuroblastoma clinical trial) has been revised from recent clinical trials and is based upon data from legacy CCG and POG clinical trials. These data suggest that patients up to 18 months of age who are diagnosed with biologically favorable stage 4 neuroblastoma share the same excellent prognosis that has previously been demonstrated for infants less than 12 months of age with non-amplification of MYCN
Children's Oncology Group
oncogene (MYCN-NA) stage 4 disease. Data from CCG 3881 and 3891 trials demonstrated a 6-year for infants months, for toddlers months, and for children 18-24 months of age at diagnosis of stage -NA neuroblastoma. In this analysis, Shimada histology also correlated with age at diagnosis and survival, with nearly all patients with favorable Shimada histology (FH) tumors being in the 12-18 month age group. Data from POG clinical trials also suggest a favorable outcome for toddlers ages 12-18 months with stage D, MYCN-NA, hyperdiploid tumors, treated on a variety of POG protocols with a 4 - year EFS of , and for patients age 6-12 months, 12-18 months, and 18-24 months, respectively, at diagnosis. Further analyses of CCG 3891 data reveal an excellent outcome for the rare patient age 12-18 months with stage 3, MYCN-NA but unfavorable Shimada histology (UH) disease. Measurement of tumor DNA index was not performed in this study. The 3 -year EFS/OS was for patients months with Stage 3, MYCN-NA and UH neuroblastoma ( ).
Based upon these data, ANBL0532 will exclude patients age 12-18 months with stage 4, MYCN-NA, hyperdiploid, and FH tumors or stage -NA, unfavorable histopathology. These patients will be treated on the intermediate-risk protocol, ANBL0531, representing a reduction of therapy in both doseintensity and cumulative dose. Of note, this clinically favorable cohort of 12-18 month old patients with stage 4 disease represents only of all stage 4 patients enrolled onto CCG3891 for whom MYCN analyses were available and only of all stage D patients enrolled onto POG trials for whom and DNA ploidy analyses were available.
In contrast, patients 12-18 months of age with stage -NA tumors characterized by either diploid DNA content (4-year EFS 66.6%) or UH tumors (6-year EFS 46%) and patients greater than 18 months of age with stage 3 tumors and unfavorable histology (5-year EFS 53%) have a less favorable outcome and support further attempts to optimize therapy. However, given that the majority of these patients treated on legacy CCG or POG trials did not receive myeloablative therapy, a further escalation to tandem transplantation may not be warranted. Therefore, patients 12-18 months of age with stage 4 MYCN-NA tumors but with diploid DNA content, UH or indeterminant histology or DNA ploidy and children greater than 18 months of age with stage -NA tumors with UH, estimated to be of all high risk stage patients, will be eligible for treatment on ANBL0532 but will be nonrandomly assigned to single myeloablative therapy. A uniformed treatment approach will allow improved analysis of outcome for this relatively rare patient cohort.
2.2 High-risk Neuroblastoma Therapy
High-risk neuroblastoma is largely chemotherapy responsive, but despite improvements in complete response rates, the majority of patients still relapse. To improve event free survival, COG and legacy CCG and POG clinical trials have intensified chemotherapy during induction and consolidation phases of therapy and added biologic agents for treatment of minimal residual disease. CCG-3891 study demonstrated that increased dose intensity of consolidation with purged bone marrow transplant and administration of the biologic agent 13 -cis retinoic acid (cis-RA) following consolidation therapy improved outcome for patients with high-risk neuroblastoma . The 8 -year EFS and OS rates for patients treated with one transplant and cis-RA ( ) are and , respectively (personal communication; K. Matthay and W.London). The recently completed COG Phase III trial, A3973, evaluated whether the use of a more intensive induction chemotherapy regimen (N7), reported by investigators at Memorial Sloan-Kettering to produce a CR/VGPR rate, will result in improved EFS and end-induction response rate when compared to CCG-3891. A3973 also tested whether the use of anti-neuroblastoma purged PBSC rescue improves EFS for patients with high-risk neuroblastoma. The results of COG A3973 indicate no difference in event-free or overall survival with the use of purged PBSC products for transplant. Feasibility and toxicity data from COG A3973, ANBL00P1 and ANBL0P2 cited below support the continued utilization of a dose intensive induction regimen and myeloablative chemotherapy consolidation regimen as the "backbone" schema as proposed in this successor study for high-risk neuroblastoma. The current COG ANBL0032 has demonstrated that immunotherapy (ch14.18 antibody plus cytokines) in addition to cis-RA improves EFS. Patients treated on ANBL0532 will be eligible for participation on ANBL0032 or ANBL0931.
2.3 Myeloablative regimens
The results of the randomized CCG-3891 protocol demonstrate that a myeloablative therapy with carboplatin, etoposide, melphalan and 1000 cGy total body irradiation (TBI) and purged autologous bone marrow rescue improves EFS for patients with high-risk neuroblastoma. The 3 -year EFS was for all patients on study, of which had stage 4 neuroblastoma. The 3-year EFS from time of assigned randomization was for autologous bone marrow transplantation (ABMT) versus for continuation chemotherapy (CC) ( ). Overall survival was not significantly different for the two regimens, perhaps partly due to use of ABMT salvage in patients who progressed after chemotherapy consolidation. An apparent advantage was seen for ABMT within strata separated either by MYCN amplification, bone marrow immunocytology or stage. More recent evaluation of outcome for those children with high-risk neuroblastoma treated on CCG 3891 revealed an overall 8-year EFS and OS rate +/- SE from the time of enrollment onto CCG 3891 of and , respectively, with a median follow-up time of patients who did not experience an event of 7.7 years ( 130 days -12.8 years) (Table 1). The 8 -year EFS from the time of randomization for patients randomized to ABMT ( ) was vs. for patients randomized to CC . The 8 -year OS for ABMT was not statistically significantly higher than for CC, vs. , (personal communication, K. Matthay and W. London). These data continue to suggest that therapy intensification diminishes risks for neuroblastoma recurrence. Based on the incremental improvement in EFS with ABMT, myeloablative consolidation has become a standard of care for patients with high-risk disease in the United States and abroad.
While TBI was used in the initial studies of autologous transplantation for neuroblastoma , the worldwide trend has been to eliminate this modality due to excessive toxicity including second malignancies. It is now clear that retrospective comparisons of TBI and non-TBI containing regimens showed no difference in patient outcomes. Smedler et al. reported a moderate general developmental delay; with motor, perceptual and cognitive defects in ten children transplanted for leukemia or neuroblastoma with TBI regimens. Children transplanted for neuroblastoma with TBI also demonstrate abnormal growth rates, in some cases associated with growth hormone deficiency. TBI regimens can also cause late effects such as cataracts, other endocrine dysfunction, and secondary malignant neoplasms. There has been no prospective comparative trial to determine whether the addition of TBI improves survival from high-risk neuroblastoma. A retrospective review of 51 patients transplanted for advanced or poorly responding neuroblastoma from the European BMT Solid Tumor Registry found no significant difference in outcome between TBI and non-TBI regimens. A separate retrospective review of 62 concurrently treated stage 4 neuroblastoma patients with and without TBI also demonstrated equivalent survival rates. The CHLA group has shown a 3-year EFS rate of in 73 patients analyzed from the end of induction therapy who were treated on protocol 91-LA6 with a non-TBI regimen of carboplatin, etoposide and melphalan with local irradiation (CEM-LI, Table 1). A preliminary comparison of the EFS of patients with stage 4 neuroblastoma over one year of age at diagnosis transplanted before progression with CEM-LI ( ) with the same cohort of patients treated with CEMTBI on CCG-321P3 ( ) showed no significant difference in outcome ( ). Overall, there were 4/73 (5%) toxic deaths in the patients transplanted before progression with CEM on the 91-LA6 protocol as compared to toxic deaths following CEM-TBI in similar patients transplanted on CCG-321P3. These data suggest that the proposed CEM-LI without TBI is at least as effective as prior TBI regimens with the caveats that sample size limited the ability to detect a statistical difference in EFS and that trend towards improvement in EFS in this historical comparison could also be attributed to general improvements in care and induction regimens used in the 91-LA-6 (personal communication, J. Villablanca).
The COG A3973 recently completed Phase III trial utilized the CEM preparative regimen based upon the results for the 91LA6 clinical trial noted above. Of the 489 eligible patients enrolled onto A3973, 368 had completed preparative regimen of carboplatin, etoposide, melphalan and autologous transplant with either purged or unpurged PBSC and reported on toxicity. Nearly of the patients developed Grade 3 or 4 stomatitis during consolidation. One patient developed Grade 3 cardiac dysfunction, and four had renal
dysfunction. Three patients developed renal failure requiring dialysis. Two of these patients had severe sinusoidal-obstruction syndrome (SOS), formerly VOD, the other had severe capillary leak syndrome with grade 2 elevation of bilirubin, but no hepatic enlargement. All 3 patients proceeded to post transplant therapy on study. In total, 12 of 368 ( ) patients have died of complications during Consolidation phase of therapy, mainly due to infectious complications ( ). There have been 20 patients reported with SOS, which resulted in death in the 3 patients. These toxicities are similar to those reported using alternative myeloablative consolidation regimens and did not meet designed stopping rules. Twoyear EFS for the tranpslanted cohort from time of study enrollment was . The 91LA6 and COG A3973 experience to date suggest tolerable toxicity following CEM-LI consolidation therapy. Comparative analysis of 91LA6, CCG 321-P3 and A3973 indicates that the CEM-LI regimen without TBI is at least as effective as prior TBI regimens. Therefore, CEM-LI will be employed in each arm of this successor Phase III study comparing single versus tandem consolidation therapy.
2.4 Tandem Myeloablative Regimens
High-risk neuroblastoma remains incurable in of patients. Further dose intensity may yield further improvements in EFS. To achieve this, tandem, or dual-cycle, transplant approaches have been developed. An initial attempt at tandem transplant using marrow support was piloted by the European Neuroblastoma Group in the LMCE2. Median time to reach the second ABMT was 3 mos. (range 2-6 mos.), and median time to ANC was 43 days (15-120). Overall survival at 2 years was in a very high-risk group of patients, but the toxic death rate was high at .
Grupp et al. explored the use of tandem transplant with PBSC support in children with high-risk solid tumors. The myeloablative regimens employed in the CHP-594/DFCI 34-DAT study were an initial regimen of etoposide, cyclophosphamide and carboplatin followed 4-6 weeks later by a second regimen of melphalan with 12 Gy of TBI. Initial data on 55 enrolled patients (ages 1 to 18 years) demonstrated the feasibility of the approach with 98 cycles of myeloablative therapy with stem cell rescue completed. Four patients who completed the first HSCT course did not complete the second, and there were three toxic deaths. Median time to neutrophil engraftment was 11 days. One patient experienced delayed engraftment but recovered following infusion of back-up PBSC. The 3-year EFS was with a median follow-up of 22 months from diagnosis. These data are substantiated by a more recent analysis of 97 consecutive children (including the 55 previously published) treated with the CHP-594/DFCI 34-DAT that shows a 3 -year PFS and OS of and ), respectively; a 5 -year PFS and OS of ( CI ) and ( CI ), respectively and a 7 -year PFS of Of the 89 patients who received at least one HSCT, the 5 -year and 7 -year PFS was and , respectively (Table 1).
Toxicity and feasibility data on tandem transplantation without TBI have been evaluated in POG 9640 and COG ANBL00P1 studies. POG 9640 enrolled 33 patients and utilized 5 cycles of induction chemotherapy followed by tandem transplantation with unpurged PBSC support. The tandem regimens were: Carboplatin, etoposide, cyclophosphamide (AT#1) followed by thiotepa and cyclophosphamide (AT#2). This regimen was found to be feasible and tolerable. There were no toxic deaths associated with the tandem regimen. A total of 21 patients underwent at least one course myeloablative therapy and 17 patients received tandem regimens. Of the 17 patients who underwent tandem transplant, 12 were alive without evidence of disease at 2 years. The 5 -year EFS and OS rates +/- standard error of the 33 patients enrolled was and , respectively. Twenty-two patients received at least one transplant, and the 5-year EFS and OS rates from time of first transplant were and , respectively (personal communication S. Grupp and W. London, MS in preparation).
The promising results of the CHOP/DFCI experience, the POG 9640 study and the 91LA6 CEM-LI study have led the COG Neuroblastoma Committee to consider intensification of CEM-LI in a tandem regimen. The feasibility and toxicity of incorporating CEM-LI in a tandem regimen has been studied in COG ANBL00P1. This limited-institution pilot study utilized five cycles of induction chemotherapy followed by tandem transplantation. The tandem regimens were Thiotepa/Cyclophosphamide followed by CEM-LI. Thiotepa/Cyclophosphamide regimen was chosen as the initial transplant based upon the known toxicity
Children's Oncology Group
profile of agents alone and in combination when delivered at myeloablative doses, the acceptable toxicity profile of the combination demonstrated in the POG 9640 tandem transplant study and the known anti-neuroblastoma activity of each agent including their ability to cross the blood brain barrier. Data obtained from a CHOP pilot tandem regimen of Thiotepa/Cyclophosphamide followed by full dose CEM (equivalent to A3973 dosing) revealed excessive toxicity following CEM. Therefore, a dose reduction of CEM agents was utilized in the ANBL00P1 protocol and will be utilized in ANBL0532. The observed toxicity following CEM in the CHOP pilot and in the COG A3973 trial suggest that CEMrelated toxicity would interfere with subsequent delivery of myeloablative chemotherapy and support the sequence of thiotepa/cyclophosphamide followed by CEM in a tandem myeloablative regimen.
Of the 41 eligible patients enrolled onto ANBL00P1, 8 patients were removed from protocol prior to consolidation. Of the remaining 33 patients, 7 received only a single transplant while 26 patients received tandem transplants. Reasons for not receiving the 2 nd transplant included progressive disease ( 1 patient), family preference (2 patients), mortality following transplant #1 ( ) and prolonged toxicity following transplant #1 ( , delayed engraftment, elevated liver transaminases, renal insufficiency). Therefore, of all patients and of those patients eligible for consolidation therapy received the tandem consolidation regimen. This compares favorably to the legacy CCG3891 trial where of patients assigned to ABMT received the intended therapy. Overall, 3 patients have experienced treatment related mortality. Twenty-two events occurred in 41 eligible patients enrolled at diagnosis resulting in 2 -year EFS from time of diagnosis of . These data support the feasibility of this tandem myeloablative chemotherapy regimen, although small numbers of accrued patients limits the ability to assess efficacy.
The summary data presented in Table 1 support the investigation of tandem transplant consolidation to improve EFS from high risk neuroblastoma. A comparison of the CCG3891 and CHP/DFCI trials, both TBI-containing trials, reveals an improvement in EFS following tandem consolidation; (CCG 3891 from time randomized to ABMT: 8-year EFS 27+/- 5% versus CHP/DFCI from diagnosis: 7-year EFS 45+/ versus CHP/DFCI from time on initial myeloablative regimen: 7 -year EFS ). A trend toward improvement in EFS following tandem transplant is even observed when comparing the highly selected cohort of CCG3891 patients who received ABMT and were randomized to receive cis-RA ( of those patients patients randomized to ABMT) versus the CHP/DFCI patients who received the intended tandem transplant ( of patients enrolled); CCG 3891: 8-year EFS versus CHP/DFCI 7-year EFS 52 +/- 11%. Furthermore, the 91LA6 data suggests that CEM provides similar EFS to the single TBI-CEM containing regimen used in CCG3891, supporting the elimination of TBI from a tandem regimen. The EFS and OS results of the various non-TBI containing tandem regimens (ANBL00P1 and POG 9640) vary between studies, reflecting the difficulty in evaluating efficacy of therapy regimens in small, non-randomized clinical trials. However, the combined data from the CHP/DFCI, the POG 9640 and the ANBL00P1 trials demonstrate that intensification of consolidation therapy with PBSC support is feasible, does not increase treatment related mortality and may decrease recurrence for patients with high-risk neuroblastoma.
Children's Oncology Group
Table 1. Comparison of Myeloablative Regimens for Treatment of High Risk Neuroblastoma
CCG3891 Overall (379 randomized)
CCG3891 ABMT (129 received ABMT)
CEM-L1 (91LA6)
CHP594/ DFCI34
POG9640
ANBL00P1
ANBL02P1
Preconsolidation Response (%CR/ VGPR)
CR + VGPR 50% (190/379)
PD=50/379
CR+ VGPR 52% (98/189)
PD=28/189
CR + V GPR 53%
CR+VGPR +PR+MR 94% (91/97)
CR+ VGPR 70% (23/33)
CR+VGPR+P 90% (37/41)
CR/VGPR 50% (13/26)
% stem cell collections
BM source
BM source
NA
NA different induction
NA different induction
NA different induction
88%
%EFS at 2y
Too early
%EFS at 3y
NA
% EFS at 5y
NA
% EFS at 7y
Too early
NA
% local relapse
9.9%
12.4%
Data not available
Data not available
Data not available
% second cancer
0.55% (3/539) 2 leuk, 1 clear cell sarcoma
0.5% (1/189)
2.8% (2/73)
2% (2/97)
3% (1/33)
Data not available
Too early
% HSCTrelated toxic deaths
4% (22/539)
7% (9/129)
5% (4/73)
5% (5/97)
0% (0/22)
6% (2/33)
6% (2/31)
1 = Data represent 8yr EFS data for CCG 3891; EFS calculated from time of enrollment at initial diagnosis; EFS calculated from time of initial HSCT for those patients who received ABMT and were randomized to receive cis-RA (CCG3891 ); 4= EFS calculated from time of initial HSCT (91LA6 ), or from time of initial HSCT who received at least one HSCT regimen (CHP594/DFCI34 , POG9640 N=22, ANBL00P1 ); calculated from time of initial HSCT for those patients who received tandem HSCT per CHP 594/DFCI34 ( ); 6= protocol only required a minimum collection of cells ; median collection cells (range ). Not available
2.5 Novel Induction Regimen
The emergence of chemotherapy-resistant tumor cells is the major obstacle to improved initial tumor response and to the cure of high-risk neuroblastoma. A marked escalation in chemotherapy dose intensity has resulted in improved initial tumor response rates. A highly dose intensive combination of cyclophosphamide, vincristine and doxorubicin alternating with cisplatin and etoposide has been studied in small numbers of patients at Memorial Sloan Kettering Cancer Center with a reported CR plus very good partial response rate (VGPR) of Unfortunately, this excellent induction response rate was not reproduced in a multi-center trial performed by the French Society of Pediatric Oncology (SFOP) where 21 of 47 patients who received the MSKCC induction regimen achieved CR at metastatic sites of disease, results that were similar to prior less dose intensive regimens. This regimen was further evaluated in over 400 patients with high risk neuroblastoma enrolled on the recently completed COG Phase III trial (A3973), demonstrating a CR plus VGPR of . Data from the A3973 and SFOP trials suggest that chemotherapy resistance remains an obstacle to the cure of high risk neuroblastoma. Further dose intensification of induction chemotherapy is limited by hematopoietic and mucosal toxicity arguing for the addition of new non-cross resistant agents to further improve tumor response rates.
2.6 Topotecan
Topotecan, 9-dimethylaminomethyl-10-hydroxycamptothecin, is a semi-synthetic derivative of camptothecin that inhibits topoisomerase I enzyme resulting in double stranded DNA breaks and cellular apoptosis. Preclinical investigations in murine xenograft models have demonstrated antitumor activity against neuroblastoma, synergistic activity when combined with alkylator and platinum anti-tumor compounds and both dosage and schedule dependency of anti-tumor activity. Anti-tumor activity demonstrated in xenograft models is maximal following daily x 5 days administration or daily x 5 days x 2 weeks at single day systemic exposure of , a systemic exposure proven to be feasible in subsequent clinical trials. Although topotecan demonstrates a non-cross resistant mechanism of action cross-resistance to etoposide, a topoisomerase II inhibitor commonly used in neuroblastoma therapy, has been demonstrated by in vitro analyses of patient-derived neuroblastoma cell lines resistant to etoposide. Patient-derived neuroblastoma cell lines established early in the clinical course (diagnosis or after induction therapy) were more likely to be sensitive to topotecan, supporting the incorporation of topotecan into Induction therapy, prior to significant etoposide exposure. In vivo, topotecan is eliminated through hepatic metabolism and renal excretion. Topotecan penetrates the CNS, a site of recurrence in neuroblastoma. Topotecan has a limited toxicity profile notable for dose-limiting myelosuppression and mild to moderate nausea, vomiting and mucositis, regardless of administration schedule. Antineuroblastoma activity has been demonstrated in both Phase I and II trials. A Phase I trial administered topotecan at dosages of day to day for 5 days to pediatric patients with recurrent tumors. Of nine patients with neuroblastoma, a partial response was demonstrated in 3 patients while stable disease occurred in one patient. A Phase II upfront window trial in patients with newly diagnosed high-risk neuroblastoma (POG-9341) identified topotecan ( day for 5 days) as an active antineuroblastoma agent with of patients achieving a complete ( ) or partial tumor response ( ) following 2 cycles of single agent therapy. In a more traditional topotecan single agent Phase II trial (topotecan day for 5 days) enrolling 37 patients with refractory neuroblastoma, complete response (CR) was achieved in 2 patients, mixed response (MR) in 5 patients and stable disease (SD) in 8 patients. These data are consistent with topotecan's anti- neuroblastoma efficacy documented in preclinical studies and support the incorporation of topotecan into upfront therapy for neuroblastoma.
2.7 Topotecan and Cyclophosphamide
Topotecan and cyclophosphamide have been successfully combined without excessive toxicity and have anti-neuroblastoma activity. A phase I trial in pediatric patients with refractory or recurrent solid tumors demonstrated the topotecan MTD as day administered over 30 minutes daily for 5 days in combination with cyclophosphamide day administered daily for 5 days. Myelosuppression, defined as Grade 4 neutropenia, was the dose limiting toxicity without additional significant toxicity observed. Anti-tumor activity was further evaluated in a Phase II trial of topotecan day x 5 days combined with cyclophosphamide day for 5 days administered to 83 pediatric patients with refractory solid tumors. Six of thirteen patients with refractory/recurrent neuroblastoma ( 5 received prior HSCT) achieved a PR ( 3 had received a prior BMT), while 2 patients achieved mixed response or stable disease. Preliminary data from POG-9642, a randomized study of topotecan (2 day for 5 days) versus topotecan/cyclophosphamide (at Phase II doses noted above) in patients with recurrent neuroblastoma demonstrate a trend toward improved CR/PR rate following topotecan/cyclophosphamide therapy ( versus 20%), decreased rate of grade 3 or 4 infectious complications and increased time to tumor progression (Frantz C., COG 9462 Public Report, 2/11/2003).
Xenograft models predict that higher systemic exposure ( ) to topotecan will improve tumor response. A topotecan systemic exposure of is predicted to be achieved by doses of 1.0 to day. Limited pediatric trials, both non-myeloablative and myeloablative, have incorporated such doses of topotecan. Repetitive delivery of a dose-intensive topotecan and cyclophosphamide regimen (MSKCC regimen), continuous infusion high-dose cyclophosphamide ( 4200 hours) combined with continuous infusion topotecan ( hours), has been administered to pediatric patients with recurrent solid tumors. Severe neutropenia was observed following all courses of therapy, with of courses complicated by fever and complicated by
bacteremia. Median duration of neutropenia was 7 days (range days); however, all patients recovered neutrophil count to and platelet to by 28 days. Nonhematologic toxicity was limited to Grade 4 hyperbilirubinemia and Grade 3 cardiac dysfunction in one patient during documented sepsis and three episodes of Grade 3 mucositis, two associated with herpes simplex infection. There were no toxic deaths. Cyclophosphamide and topotecan doses of day for 3 days and 1.25 to 4 day for 5 days have been successfully combined with melphalan as a myeloablative regimen for adult patients with advanced ovarian cancer. Non-hematopoietic toxicity was limited to mucositis with maximum of Grade 2 toxicity occurring in 16 of 53 patients treated.
The limited toxicity profile following dose-intensive administration of topotecan and cyclophosphamide support the potential feasibility for its incorporation into an upfront induction regimen. To this end, COG ANBL02P1 examined the feasibility of adding dose intensive topotecan and cyclophosphamide onto the N7 backbone chemotherapy administered in COG A3973 study. Escalated doses of topotecan (1.2 day for 5 days) and cyclophosphamide ( day for 5 days) (T/C) replace the initial 2 cycles of A3973 induction chemotherapy allowing incorporation of topotecan while maintaining dose intensity of known active agents in the treatment of neuroblastoma (see Table 2). The topotecan dose of day days was chosen based upon a predicted topotecan AUC ( *hr) that is within the range known to produce tumor response in murine xenograft models and the ability to administer with cyclophosphamide. Chemotherapy cycles were scheduled every 21 days, PBSC harvest occurred after T/C cycles and surgical resection of residual primary tumor after cycle 5. Thirty-one patients, 3 with INSS Stage 3 and 28 with Stage 4 were enrolled. Pharmacokinetically guided topotecan dosing (target systemic exposure of AUC determined by single day topotecan lactone levels) demonstrated the ability to achieve the target AUC in of patients during T/C Cycle 1 and in of patients during T/C Cycle 2. Furthermore, the median topotecan dose required to achieve the targeted AUC during Cycle 1 and Cycle 2 was day and day , respectively and support the use of day dosing in the ANBL0532 induction. Successful PBSC harvest as defined by a minimum of CD34 cells/kg was achieved in patients for whom data are available; median harvest of cells/kg (range 2.24-548) collected over a median of 1 day (range 1-3 days). A PBSC harvest of CD34 cells was achieved in 23 patients ( ). One patient was harvested after Cycle 5 due to physician discretion and achieved a PBSC collection of CD34 cells/kg. Only one patient required 3 days of apheresis but achieved a total PBSC collection of CD34 cells/kg. All PBSC collections were free of tumor contamination using an immunocytochemical detection assay. There were no dose limiting toxicities during Cycles 1 and 2 of induction and no induction related moralities. The majority of patients ( ) experienced Grade 3 or 4 hematopoietic toxicity. Documented infection occurred in patients during T/C cycles compared to of patients during subsequent induction cycles on ANBL02P1 or patients during initial 3 cycles of the MSKCC induction on COG A3973. The intended dose intensity of all chemotherapy agents was maintained in 30 of 31 patients. One patient required a dose reduction of chemotherapy doses during Cycle 6 induction due to prolonged myelosuppression. Complete response or partial response was achieved in of patients following 2 cycles of T/C. End-induction response rate (CR/VGPR) was ( ), 1 patient developed progressive disease following completion of induction but prior to initiating consolidation therapy.
In summary, this dose intensive cyclophosphamide and topotecan containing induction regimen was well tolerated with expected and reversible toxicities. Dose intensity of standard induction chemotherapy agents was not limited by the addition of dose-intensive topotecan. The ability to achieve sufficient PBSC harvest with in vivo tumor purging was demonstrated. Together these data support the investigation of efficacy of this novel induction regimen in ANBL0532.
Children's Oncology Group
Chemotherapy Dose Intensity - Comparison of Therapeutic Regimens
TABLE 2. Comparison of Induction Regimen Chemotherapy Dose Intensity
Drug
CCG 3891 15 weeks
POG 9341 15 weeks
COG ANBL00P1 15 weeks
COG A3973 18 weeks
COG ANBL0532 18 weeks
Oxazophosphorine equivalents+
500
434
700
933
688
Doxorubicin
7.5
10
8
16.6
8.3
Platinum equivalents#
15
29.9
27.3
22.2
22.2
Etoposide
50
110
100
66.7
66.7
Topotecan
0
0
0
0
0.67
Dose intensities are calculated as week administered over the entire induction regimen.
Oxazaphosphorine = Cyclophosphamide + Ifosfamide. The dose intensity of ifosfamide is calculated in cyclophosphamide equivalent using a conversion of 4 mg Ifosfamide is biologically equivalent to 1 mg of cyclophosphamide.
# Platinum equivalents Cisplatin + Carboplatin. The dose intensity of carboplatin is calculated in cisplatin equivalent using a conversion of 4 mg carboplatin is biologically equivalent to 1 mg of cisplatin.
2.8 Local Radiation Therapy
For stage III and IV neuroblastoma, local relapse is a major component of treatment failure in several published series, all of which included radiation delivered to the site of primary disease. As systemic therapy for high-risk neuroblastoma becomes more aggressive, response rates improve, and survival increases, local control becomes a formidable problem. This is evident in a recent analysis of CCG 3891, consisting of chemotherapy, primary surgery, 10 Gy of external beam radiation therapy (EBRT) to gross residual disease, followed by randomized assignment to continuation chemotherapy (CC) or autologous bone marrow transplantation (ABMT). ABMT patients received 10 Gy of total body irradiation (TBI). Estimated 5-year locoregional recurrence rates were and for CC and ABMT patients, respectively . Results of this trial revealed that for patients who received 10 Gy of EBRT to the primary, the addition of 10 Gy of TBI and ABMT decreased local recurrence compared with and . This indicates that in combination with EBRT to the primary tumors site, the addition of 10 Gy of TBI as a component of high dose chemotherapy with ABMT improved local control compared with CC without TBI. Several single institution reports further support the administration of pre-chemotherapy or pre-surgery primary tumor volume and regional lymph nodes primary site radiation therapy ( 21 Gy) results in improved local tumor control. Based upon these data, the currently completed COG A3973 delivered 21.6 Gy EBRT to pre-surgical primary tumor volume following recovery from consolidation myeloablative therapy.
Several analyses of outcome for high-risk neuroblastoma suggest that current local therapy for incompletely resected patients is inadequate. Wolden et al. reported on a series of patients with stage 4 neuroblastoma receiving 1.5 Gy twice a day to 21 Gy to pre chemotherapy, pre-surgery primary tumor volume and regional lymph nodes. The actuarial locoregional control rate at 5 years was . A recent update of this experience reported a probability of primary-site failure among 99 patients, most of whom ( 92 patients) had no evidence of disease in the primary site at the time of irradiation. Among seven patients with disease at the primary site at the time of irradiation, three had disease that recurred locally. EBRT did not increase acute toxicity, except for increased total parenteral nutrition administration.
Published studies have sought to assess the appropriate radiation dose to achieve local control in patients with neuroblastoma. A dose response to radiation is supported by several publications; however, these publications describe results in stage III disease or do not incorporate biological prognostic markers.
Children's Oncology Group
In the analysis of radiation therapy in CCG 3891, in combination with EBRT to the primary tumors site, the addition of 10 Gy total body irradiation (TBI) as a component of autologous bone marrow transplant (ABMT) improved local control compared with continuation chemotherapy (CC) without TBI. In addition, a dose response to radiation administered to the primary tumor site is supported by the patients in this study that received 20 Gy for extra-abdominal primary tumors. Of 36 patients with extraabdominal primaries, 6 patients received 20 Gy EBRT (2 also received TBI) while 30 patients received no EBRT (10 of these received TBI). Local relapse rates at 5 years were and for patients with and without EBRT, respectively Jacobson et al. reported on the experience at the University of Florida, using radiation doses up to 45 Gy. The authors noted no evaluable complications related to radiation alone, but rather related more commonly to the tumor itself or surgical resection. Investigators at the University of Utah performed a similar analysis of radiation dose response for neuroblastoma. The authors observed local failures after doses up to 45 Gy in children older than three years. Although authors of both studies state that doses lower than 45 Gy may be adequate for some patients with neuroblastoma, the vast minority of patients in these studies had high-risk disease and therefore no firm conclusions can be drawn regarding the appropriate dose to the primary site in patients with high-risk disease. In fact, a recent abstract from the University Children's Hospital in Cologne, Germany, reported that 36 Gy of external beam radiation therapy (EBRT) administered to patients with incompletely resected primary tumors could compensate for "the disadvantage of incomplete response to induction chemotherapy." In this report of the NB97 trial for patients one year of age or older with stage 4 neuroblastoma, patients with isolated localized residual disease had improved outcome following EBRT ( 3 -y-EFS 100%, 3-y-OS 100%) compared those not receiving EBRT despite residual tumor tissue ( 3 -yEFS , p<0.001; 3-y-OS , p<0.001).
Given the high rates of local recurrence, particularly following an incomplete surgical resection, and the tolerable toxicity of EBRT evident in multi-institutional trials, patients enrolled onto ANBL0532 who have an incomplete surgical resection of the primary tumor will receive 21.6 Gy EBRT to the postinduction chemotherapy, pre-operative primary tumor volume and an additional boost of 14.4 Gy EBRT to the gross residual volume (total dose 36 Gy to gross residual tumor volume). The additional dose of 14.4 Gy to gross residual disease is unlikely to result in significantly increased toxicities. In CCG 3891, the administration of EBRT was not associated with enhanced toxicities with the exception of parenteral nutrition requirement, which occurred in and of patients who did and did not receive EBRT, respectively . Furthermore, 36 Gy is well within normal tissue tolerance for most organs within the field of radiation. Kidneys and liver will likely prove to be the dose-limiting structures and the same guidelines for maximal doses to normal structures will be followed, as is standard in all current neuroblastoma protocols. It should also be noted that the boost for patients with incomplete resections will deliver an additional 14.4 Gy (from 21.6 Gy to 36.0 Gy) only to regions of gross residual disease, a target that generally is substantially smaller than the original pre-operative tumor volume that receives 21.6 Gy.
2.9 Surgery
Biopsy at initial diagnosis is necessary to obtain adequate tissue for biologic studies and enrollment on the Neuroblastoma Biology Protocol. Resection of the primary tumor and bulky metastatic disease is usually necessary to achieve CR or VGPR after induction chemotherapy. A retrospective analysis of high risk neuroblastoma patients enrolled onto CCG 3891 demonstrated improved resectability of primary tumor after initial chemotherapy and revealed a trend toward improved survival for those patients who underwent gross total resection of primary tumor, 5-year EFS and , respectively Acute surgical complication rates and characteristics were independent of timing of primary tumor resection. Based upon these data, patients enrolled onto the recently completed A3973 underwent delayed surgical resection of the primary tumor after 5 cycles of chemotherapy. Tumor biopsy at diagnosis was required to obtain adequate tissue for assessment of biologic characteristics. ANBL0532 will utilize this treatment strategy and prospectively evaluate if complete primary tumor resection is predictive of local control and EFS in high risk neuroblastoma patients.
Early and late postoperative complications are known to occur following extensive tumor resection for neuroblastoma. These complications may include adhesive bowel obstruction, chylous leak, ipsilateral renal injury, atrophy or loss, and chronic diarrhea. Group wide studies have regularly sought data on intraoperative and 30 day postoperative complications. Adkins et al. reported normal organ removal in of high risk neuroblastoma patients who underwent complete resection of primary tumor. There was no statistical difference in acute intra-operative or peri-operative complications, including risk for hemorrhage, renal injury, wound complication or acute bowel obstruction, regardless of extent of surgical resection. There is potential for bias in this study, given the retrospective collection of data. Extended observations are necessary to detect late postoperative complications, as they may relate to the extent of resection and subsequent surgical and radiotherapeutic efforts to achieve local control. Specifically, extended follow up will be necessary to detect the occurrence of adhesive bowel obstruction and renal atrophy related to surgically induced ischemic events while the risk for chylous leak or diarrhea may be related to extensive retroperitoneal dissection. Rates of these complications should be correlated with the surgical and radiotherapeutic efforts in obtaining local control as they may impact on future management decisions.
2.10 Neurologic Outcome of Patients with Epidural Neuroblastoma
Epidural or intradural extension of tumor occurs in approximately of patients diagnosed with neuroblastoma, and may occur with or without neurologic impairment. Epidural neuroblastoma with spinal cord compression is an oncologic emergency that warrants immediate intervention. However, the choice of emergent therapy remains somewhat controversial, with pediatric oncologists favoring chemotherapy and neurosurgeons favoring laminectomy with surgical decompression. There are several retrospective reports of similar neurologic outcome for patients treated with chemotherapy versus laminectomy, and therefore emergent chemotherapy has become a preferable alternative to laminectomy in most cases, in an effort to diminish the devastating late orthopedic effects of laminectomy in infants or young children. Radiation therapy has generally been reserved for situations of progressive or persistent neurologic dysfunction despite chemotherapy and laminectomy.
This study will prospectively characterize multiple aspects of neurologic disability at diagnosis and during the follow-up period in an effort to systematically study the neurologic outcome of patients with primary neuroblastoma tumors with intraspinal extension (see Appendix III). A multidisciplinary evaluation at the time of diagnosis, including oncology, neurosurgery, and radiation oncology, is strongly encouraged. It is the intent of this study that patients with clinical signs of spinal cord compression will be treated with primary chemotherapy, unless the multidisciplinary evaluation determines that surgical decompression would be more appropriate. Radiation therapy should be reserved for patients whose neurologic status deteriorates despite chemotherapy and surgical decompression.
2.11 Maintenance Therapy
The presence of minimal residual disease despite achievement of maximal response to induction and consolidation therapy results in continued risk for neuroblastoma relapse. This was demonstrated in the CCG-3891 study where the benefit of additional non-cross resistant therapy, 13-cis Retinoid acid (cisRA) administered following myeloablative consolidation therapy was observed. Patients with newly diagnosed high-risk neuroblastoma enrolled in CCG3891 were randomized at week 34 of therapy (following either chemotherapy or ABMT consolidation) to no further therapy versus cis-RA for six months. Cis-RA or isotretinoin is a synthetic retinoid derived from the naturally occurring all transretinoic acid by modification of the terminal carboxyl group. When neuroblastoma cell lines are exposed to all-trans-retinoic acid (trans-RA) or cis-RA in vitro, they exhibit decreased proliferation, decreased expression of the MYCN oncogene, and morphological differentiation. Growth arrest and differentiation in response to cis-RA have been observed in neuroblastoma cell lines initiated from tumors at the time of progression after chemoradiotherapy, suggesting that resistance to cytotoxic chemotherapy does not induce resistance to cis-RA. cis-RA was well tolerated in a pediatric Phase I trial using an intermittent administration schedule of twice daily administration for 14 days followed by 14 days with no therapy. cis-RA toxicities are generally mild; consisting primarily of chelitis, dry skin, and hypertriglyceridemia, with hypercalcemia seen at higher doses. A significant difference in the three
year EFS from the time of this randomization in the patients receiving cis-RA ( ) versus those with no further therapy ( ), with p value of 0.027 in a test of proportions. This advantage for EFS was seen in subgroups by prior randomization, such that the best overall EFS was for patients treated with ABMT with cis-RA ( EFS), second was ABMT without cis-RA ( ), third was chemotherapy consolidation with cis-RA ( ), and fourth was chemotherapy without cis-RA ( ).
Despite the use of cis-RA, greater than of children will develop recurrent neuroblastoma. To further improve outcome, the efficacy of novel, anti-neuroblastoma targeted immunotherapy to eliminate minimal residual disease has been evaluated. Anti-gangliosidase (GD2) monoclonal antibody ch14.18 has shown preclinical and early clinical activity against neuroblastoma, which was enhanced when combined with GM-CSF or IL2. COG ANBL0032 was designed to determine if adding ch14.18 + GM-CSF + IL2 to standard therapy of isotretinoin after intensive multimodality therapy improved outcome for high-risk neuroblastoma patients. High-risk neuroblastoma patients who responded to induction therapy and stem cell transplant were randomized to 6 cycles of isotretinoin (standard therapy) or 6 cycles of isotretinoin with 5 concomitant cycles of ch14.18 combined with GM-CSF or IL2 in alternating cycles (immunotherapy). An intent-to-treat randomized comparison (Lan-DeMets interim monitoring, cumulative alpha ) was performed for event free survival (EFS). A total of 226 eligible patients were evenly randomized to standard or immunotherapy. Immunotherapy was associated with grade pain, vascular leak syndrome and hypersensitivity reactions in and of patients, respectively. With of expected events observed, the study met criteria for early stopping for efficacy. Median follow-up was 2.1 years. Two-year EFS estimates were for patients randomized to immunotherapy versus for standard therapy ( ). OS was also superior ( , unadjusted for interim looks) for immunotherapy: versus isotretinoin alone . Moreover, for the major subgroup of patients year of age with stage 4 disease, EFS was significantly greater ( 0.02 ) for the immunotherapy group ( ), as compared to isotretinoin alone ( ). (*2-year estimates) (Alice Yu; personal communication, COG ANBL0032 study report).
Immunotherapy consisting of ch14.18 with GM-CSF and IL2 significantly improves outcome for highrisk neuroblastoma patients. Ch14.18 remains under clinical investigation through COG ANBL0032 and ANBL0931 protocols to further evaluate efficacy and toxicity. Given the significant improvement in survival from high risk neuroblastoma following antiGD2 antibody therapy, patients enrolled onto ANBL0532 should be encouraged to participate in actively enrolling clinical trials of ch14.18 antibody, (i.e. ANBL0032 or ANBL0931). Patients who are not eligible to receive ch14.18 antibody therapy or decline immunotherapy will remain on ANBL0532 and receive six months of cis-RA post ASCT based on the significant improvement in EFS with cis-RA documented by CCG3891 trial.
2.12 Biologic Correlative Studies
2.12.1 Enrollment on COG ANBL00B1
Enrollment on COG ANBL00B1 (Neuroblastoma Biology Study) is an eligibility requirement for ANBL0532 as assignment to clinical risks groups and different COG neuroblastoma therapeutic clinical trials is dependent upon accurate histologic and biologic diagnosis. Although submission of paraffinembedded tissue is acceptable for enrollment onto ANBL00B1, the COG Neuroblastoma committee encourages the submission of fresh or snap-frozen tissue, peripheral blood, or bone marrow specimen. The acquisition of these primary tumor specimens will enable additional correlative biologic studies. Enrollment on both COG ANBL0532 and ANBL00B1 has the significant advantage of allowing the correlation of novel biologic parameters with a clinically well-characterized (including treatment and outcome) cohort of patients.
212.2 Cyclophosphamide Pharmacogenomics in Pediatric Patients
Although the efficacy of cyclophosphamide is well established in NB patients, considerable inter-patient variability exists in response and toxicity. Variability in cyclophosphamide metabolism may result in such clinical variability. Cyclophosphamide dosing based on body weight ( ) or BSA ( )
results in a wide area under the concentration-time curve (AUC) range of the parent drug, and an even greater variability in the AUC of its metabolites. Children have a faster clearance of cyclophosphamide relative to adults, although the clinical significance of this difference in disposition is unclear. Recent studies have revealed an association between the AUC of cyclophosphamide and its metabolites to the risk of recurrence in children receiving standard dose cyclophosphamide (i.e., ) for treatment of B-cell non-Hodgkin lymphoma.
The pharmacokinetics of cyclophosphamide and its metabolites HCY and CEPM were evaluated in 18 of the 31 patients with newly diagnosed high-risk neuroblastoma enrolled in COG ANBL02P1. Median patient age was 2.5 years (range: 0.9-9.4).
Table 3: Pharmacokinetics of CY/topotecan
Dose 1
Dose 4
AUC
529 (332-1042)
329 (168-922)
31.9 (12.2-102.3)
42.4 (16.3-81.5)
27.3 (8.3-69.2)
30.4 (10.4-92.4)
AUC HCY/CY Ratio
0.058 (0.031-0.181)
0.113 (0.039-0.286)
Median (range)
The median clearance was (range: ) after dose 1 and 4.35 (1.55-8.54) after dose 4, consistent with previous reports Topotecan did not appear to effect cyclophosphamide clearance. Similar to our observations in patients receiving Cyclophosphamide/TBI, there was considerable interpatient variability in the AUC of cyclophosphamide with even greater interpatient variability in exposure to the metabolites. After dose 1, variability of the AUC of cyclophosphamide ( 3.1 fold), HCY ( 8.4 fold) and CEPM ( 8.3 fold) was comparable to that measured in cyclophosphamide/TBI patients (i.e., 3.3, 7.8 and 16.1 fold, personal communication J. McCune). Auto-induction of cyclophosphamide clearance was observed in 14 of the 15 children for whom cyclophosphamide AUC was available after doses 1 and 4, again similar to other reports. The and increased in four and three children, respectively.
Multivariate analysis was conducted to evaluate for determinants of AUC of cyclophosphamide, HCY and CEPM. Two of the 21 children received cyclophosphamide ; their AUCs were normalized to cyclophosphamide to allow for inclusion in this multivariate analysis. Multivariate regression analysis indicated that age, weight and body surface area explained only of the variability in CY clearance after dose 1. Furthermore, the AUC of cyclophosphamide and its metabolites were not consistently correlated.
Cyclophosphamide pharmacokinetic assessments are unlikely to be used clinically given the variability in cyclophosphamide metabolism and the resource intensity of cyclophosphamide pharmacokinetic evaluations. Alternatively, genetic polymorphisms in genes involved in cyclophosphamide metabolism may correlate with cyclophosphamide pharmacokinetics and may predict toxicity and anti-tumor activity of the ANBL0532 cyclophosphamide containing induction regimen. Cyclophosphamide is activated by several cytochrome P450 enzymes which have genetic polymorphisms, specifically CYP3A4/5, CYP2C19, CYP2C9 and CYP2B6. CYP2B6 and alleles have been associated with either altered activation of HCY or clinical outcome. Their frequencies in Caucasians are and , respectively. Several genetic polymorphisms in CYP2C9 have functional consequences on the enzyme's activity; CYP2C92 and CYP2C93 are the most common variants. In Caucasians, of individuals are homozygotes for CYP2C91, 22% are heterozygous or homozygous for CYP2C92 and 13% carry CYP2C9*3. The expression of these alleles is less common in African-Americans or Chinese (<2% -
Children's Oncology Group , respectively). The CYP2C92 allele is characterized by an arginine to cysteine exchange at position 144 (Cys144 - Ile 359), while in CYP2C93 isoleucine 359 is changed to leucine (Arg144 Leu359). Both CYP2C92 and CYP2C93 are non-null alleles which code for functional protein with reduced levels of enzyme activity. CYP2C92 carriers have a moderate reduction in activity, while the reduction in activity is most marked for CYP2C93. GSTA1, the predominant GST involved in the metabolism of cyclophosphamide, exhibits a polymorphism that influences its hepatic expression. GSTA1B, which is several linked single nucleotide polymorphisms in the proximal promoter region of the GSTA1 gene, has reduced levels of GSTA1 enzyme. The expression of GSTA1B does not differ by ethnic group; GSTA1*A/B occurs in and GSTA1B/*B occurs in of a population of breast cancer patients.
ANBL0532 will assess whether host genetic polymorphisms in CYP2B6, CYP2C9 and GSTA1 genotypes involved in the metabolic activation and clearance of cyclophosphamide are correlated with Cyclophosphamide/Topo associated toxicity and tumor response. Prior studies have established genotype-phenotype relationships for the CYP2B6, CYP2C9 and GSTA1 genes for both in vitro analyses and in metastatic breast cancer patients receiving high dose cyclophosphamide as a component of a HSCT preparative regimen. The pharmacology laboratory at the Fred Hutchinson Cancer Research Center is currently conducting an analysis of CYP2B6, CYP2C9, CYP3A5, ABCC2, GSTA1, GSTM1, GSTT1 genetic polymorphisms in 147 patients who received CY in combination with TBI in preparation for HSCT. Preliminary analysis suggests that polymorphisms in CYP2B6, CYP2C9, and GSTA1 are key enzymes associated with toxicity (personal communication, J. McCune). An exploratory analysis of covariance (ANCOVA) was conducted using limited ANBL02P1 data. Age was used as a co-variate for three genes (GST, CYP2C9, CYP2C19) upon the following endpoints after the first cyclophosphamide dose (i.e., dose 1): AUCCY, CY clearance, AUCHCY, AUCCEPM. CYP2C9 was the only gene that trended towards statistical significance with AUCCY, cyclophosphamide clearance ( ), AUCHCY , AUCCEPM ( ).
2.12.3 13-cis Retinoic acid Pharmacokinetics
The variability of cis-RA continuous steady state (CSS) plasma levels and/or genetic variations in retinoic acid metabolic enzymes may correlate with event-free survival or systemic toxicity (CTC Grade skin, hypercalcemia and hepatic toxicity) in high-risk neuroblastoma patients. Pharmacokinetics studies performed on patients in the Phase I trial of cis-RA showed significant interpatient and intrapatient variability in peak serum levels (mean peak serum cis-RA concentration: ; trough concentration: ). It was noted that peak serum concentrations above were associated with a higher incidence of grade 3 and 4 toxicities in patients. cis-RA may be subject to first-pass metabolism and subsequent plasma (and tumor) concentrations will depend on the rate of metabolism to its inactive 4-oxo metabolite. Preliminary data from a pilot study in the UK (UKCCSG Study PK 2000 08 ) has shown that plasma concentrations of 4-oxo-13-cis-RA can accumulate to exceed those of the parent compound. A - fold variation in 4-oxo-13-cis-RA peak plasma concentrations was found. It is proposed that increased levels of this metabolite in vivo may lead to a diminished efficacy of cis-RA.
A number of cytochrome P450 (CYP) enzymes have been identified as playing a role in the metabolism of cis-RA. CYP2C8 is most important in terms of activity and level of expression in the liver. Genetic polymorphisms in the CYP2C8 gene have been described which result in a lower rate of paclitaxel metabolism and are commonly seen in a Caucasian population. Thus, genetic variation in CYP2C8 activity could underlie individual differences in cis-RA metabolism and bioavailability. The fetal isoform CYP3A7, which is expressed post-natally in a significant number of individuals, also metabolizes cis-RA . The expression of CYP3A7, and thus the contribution of this isoform to cis-RA metabolism, may be predicted by genotyping in a pediatric population. A further aspect of cis-RA metabolism is glucuronidation, both of the parent drug and of 4-hydroxy-metabolites. This conjugation may be mediated by UGT1A1 or UGT2B7, both enzymes that are subject to genetic polymorphisms. Results from this study will provide an insight into whether modulation of cis-RA dosing according to blood levels achieved and/or genotype, could be used in future studies to optimize the treatment of high-
Children's Oncology Group
risk neuroblastoma. This study will be conducted in collaboration with the United Kingdom Children's Cancer Study Group (UKCCSG).
2.12.4 Topotecan Pharmacokinetics
Many individual pharmacokinetic studies of topotecan in children with cancer have been conducted, and results have shown wide interindividual variability in topotecan clearance. Due to the inherent variability in topotecan disposition, investigators have used pharmacokinetically guided dosing to individualize the topotecan dosage for each patient based upon their systemic clearance and the goals of the clinical trial. The response rate for this approach has been promising. Although pharmacokinetically guided topotecan is a promising approach to dosing topotecan, it has limitations and it is important to find ways to simplify the current pharmacokinetic dosing strategy.
One approach would be to use a dosing model based upon patient specific covariates identified in a population pharmacokinetic analysis. Such an analysis of topotecan lactone pharmacokinetics has been conducted in a large pediatric patient population, which includes patients with many different diagnoses. The covariates identified in this analysis included BSA, age, phenytoin coadministration, calculated glomerular filtration rate, and serum creatinine. However, for the current application the patient population will be limited to children with high-risk neuroblastoma and only total topotecan (combination of lactone and carboxylate forms) will be available for pharmacokinetic analysis. Thus, a more limited population pharmacokinetic analysis was undertaken of the ANBL02P1 total topotecan plasma concentration-time data. In this analysis, the only patient covariate identified as significantly related to topotecan total clearance was sex with females having a lower clearance. Clearly this covariate will not be very informative in a dosing model. Thus, it would be helpful to gain additional covariate data in this patient population, and relate that to total topotecan clearance determined using a limited sampling model. Ultimately, data collected in this Phase III clinical trial will be used to derive a dosing algorithm for topotecan in children with high-risk neuroblastoma that will either not need plasma topotecan concentrations or at most use only one plasma concentration. By using this dosing model, the interpatient variability in topotecan systemic exposure can be minimized, and the full therapeutic efficacy of this compound can be realized.
2.12.5 Cellular Immune Responses to Neuroblastoma
Despite low or absent expression of MHC class I, T cells capable of recognizing neuroblastoma cells and acting as effector cells have been described. Survivin has the potential to be a relevant tumor antigen in neuroblastoma, as it is widely expressed and expression is inversely correlated with prognosis. Moreover, as a key inhibitor of apoptosis, survivin may be required for tumor growth and development, making its potential loss as a means of escape of the tumor from immune surveillance difficult. We hypothesize that cytolytic T lymphocytes (CTL) directed against survivin+ targets will be detectable in the blood of neuroblastoma patients. Additionally, these CTL will be expanded by exposure to two different preparations of antigen presenting cells (APCs). One APC type will be transfected with survivin mRNA, providing surviving peptides to the MHC of the APC, while the second APC type will be loaded with neuroblastoma RNA. The APC used in our studies has been an activated B cell, referred to as a CD40-B. Relevant prior work from the Grupp and Vonderheide labs show that: 1) CD40-B cells serve as efficient APCs, inducing antigen-specific and anti-tumor CTL. 2) Large numbers of CD40-B APCs can be grown from small volumes of peripheral blood, as opposed to dendritic cells, which require pheresis to collect in adequate numbers. 3) RNA transfection efficiently transduces CD40-B APC, allowing expansion of tumor-specific CTL from patient blood samples even in a tumor like neuroblastoma in which tumor antigens are not known. 4) Neuroblastoma expresses survivin. 5) Survivin-specific CTL can be detected at diagnosis in up to of neuroblastoma patients and expanded using either survivintransfected CD40-B or CD40-B transfected with neuroblastoma RNA. Iinitial studies demonstrate that T cells active against survivin+ targets emerge whether survivin RNA or whole tumor RNA is used as the antigenic stimulus. 6) Survivin-specific CTL secretes IFN-gamma in response to tumor and effectively lyse survivin+ neuroblastoma targets. 7) CD107a is a marker of CTL function and can also be used to demonstrate that CTL specific for the immunodominant epitope of survivin are responsible for antineuroblastoma killing activity.
These data point to survivin as an important tumor antigen in neuroblastoma, show feasibility of using tumor RNA to provide an antigenic payload to APCs and confirm that neuroblastoma is a tumor susceptible to immune recognition. Data revealing tumor RNA-loaded APC produce tetramer+ CTL directed against survivin that are capable lysing autologous tumor provides key evidence of the importance of survivin in neuroblastoma. These results need to be confirmed in the larger number of samples available from the patients on this clinical trial. These experiments will allow us to further characterize survivin as a potential tumor antigen in neuroblastoma.
2.12.6 Cellular Immunity after Myeloablative Chemotherapy
Increasing intensity of therapy has the potential to increase extent and duration of immune suppression, especially of adaptive immunity. This was apparent in the CHOP/DFCI tandem HSCT experience where a combination of tandem HSCT, use of TBI and, potentially, use of CD34 selection of PBSC produced significant T cell immune suppression. We hypothesize that patients on the tandem HSCT arm will experience slower T cell recovery, specifically of CD4+ T cells. Enumeration of CD3, CD4 and CD8 cells post HSCT is available at all FACT-approved HSCT centers, and is part of post-HSCT routine follow up at many centers. There is also value in following CD4 recovery to guide the level of viral surveillance and isolation precautions that may or may not be required for an individual patient. Thus, we propose to perform these assays at 1,3 and 6 months post-HSCT, comparing T cell recovery across arms of the study. These assessments will provide a valuable baseline for studies that may utilize either of the ANBL0532 regimens, especially in the context of immunotherapies that may require functional T cell recovery.
2.12.7 Minimal Residual Disease
Despite initial response to therapy, the majority of children with high-risk neuroblastoma will develop recurrent or progressive disease. More sensitive methods for the detection of minimal residual disease (MRD) are needed. Several reports demonstrated the high sensitivity ( to normal cells) of immunocytological assays in bone marrow, bone marrow autograft and peripheral blood samples. Quantitative levels of bone marrow infiltration using immunocytological assays have been predictive of outcome for patients with stage 3 and 4 disease. . More recently amplification of tissuespecific mRNA by reverse transcriptase polymerase chain reaction (RT-PCR) has been used to detect minimal disease burden (reviewed in ). Investigators have employed an RT-PCR assay, using both tyrosine hydroxylase (TH) and the neuronal gene (PGP 9.5) which has a sensitivity of 1 tumor cell per million hematopoietic cells. Although IC and RT-PCR appear to be ideal tools for monitoring MRD, reaching a sensitivity of one tumor cell detected in to normal cells, their introduction into clinical management of children with neuroblastoma has been slow. ANBL0532 will evaluate the clinical relevance of tumor cells detected by these techniques. Immunocytology and RT-PCR studies will be used on bone marrow, peripheral blood, and PBSC collections to examine the question of tumor contamination. The incidence of RT-PCR positivity in stem cell products infused will be prospectively monitored. Furthermore, the incidence of RT-PCR positivity in bone marrow and peripheral blood will be compared following consolidation therapy between single verus tandem myeloablative therapy.
2.13 Gender and Race Differences
In CCG-3891, stage at diagnosis and EFS were similar between genders, however there was a trend for males to have better EFS than females on the ABMT arm of the study. There was a significant interactive effect, in which the benefit of ABMT was restricted to males. No differences in outcome by gender were observed in the earlier 321P3 transplant study. There were no differences on CCG-3891 with respect to gender, regarding the effect of 13-cis-RA on EFS. Data from CCG-3891 also showed a lower overall EFS for African Americans versus Whites or Hispanics. African Americans had equivalent outcome on the ABMT arm, but lower EFS if treated on the chemotherapy arm in comparison with Whites. No ethnic differences were seen in the outcomes of patients treated with cis-RA.
Patients will undergo collection of peripheral blood stem cell product following 2 cycles of induction chemotherapy. The ANBL0532 study has previously mandated immunocytology assessment of peripheral blood stem cell (PBSC) product for tumor contamination. The laboratory at Children's Hospital of Los Angeles (CHLA) that has previously performed the immunocytology assay for CCG 3891, COG A3973 and COG ANBL0532 no longer has CLIA certification and no validated CLIAapproved alternative laboratory testing exists.
ANBL0532 Amendment #2 will NOT REQUIRE immunocytology assessment of PBSC for tumor contamination. The amendment to this study will also remove mandatory PBSC specimen shipment to CHLA for immunocytology testing. Data from the most recent generation of COG high-risk neuroblastoma trials show that tumor contamination is rare and is not an independent prognostic determinate of outcome. COG A3973 determined tumor cell contamination of PBSC by immunocytologic assessment in only 5 of 487 ( ) of PBSC products collected after 2 cycles of induction chemotherapy. More importantly, COG A3973 demonstrated no benefit from anti-tumor purging of PBSC product using the same antibodies used for immunocytologic detection and support the conclusion that tumor recurrence is not directly related to tumor cell re-infusion from the PBSC product. Taken together, the COG neuroblastoma committee has concluded that there are no data to support the clinical utility of immunocytological testing of PBSC products for patients with high-risk neuroblastoma.
3.0 STUDY ENROLLMENT AND PATIENT ELIGIBILITY
3.1 Study Enrollment
3.1.1 IRB Approval
Local IRB/REB approval of this study must be obtained by a site prior to enrolling patients. Sites must submit IRB/REB approvals to the NCI's Cancer Trials Support Unit (CTSU) Regulatory Office and allow 3 business days for processing. The submission must include a fax coversheet (or optional CTSU IRB Transmittal Sheet) and the IRB approval document(s). The CTSU IRB Certification Form may be submitted in lieu of the signed IRB approval letter. All CTSU forms can be located on the CTSU web page (https://www.ctsu.org). Any other regulatory documents needed for access to the study enrollment screens will be listed for the study on the CTSU Member's Website under the RSS Tab.
IRB/REB approval documents may be faxed (1-215-569-0206), emailed (CTSURegulatory@ctsu.coccg.org) or mailed to the CTSU Regulatory office.
When a site has a pending patient enrollment within the next 24 hours, this is considered a "Time of Need" registration. For Time of Need registrations, in addition to marking your submissions as 'URGENT' and faxing the regulatory documents, call the CTSU Regulatory Helpdesk at: 1-866-651CTSU. For general (non-regulatory) questions, call the CTSU General Helpdesk at: 1-888-823-5923.
3.1.2 Patient Registration
Prior to enrollment on this study, patients must be assigned a COG patient ID number. This number is obtained via the eRDE system once authorization for the release of protected health information (PHI) has been obtained. The assigned COG patient identification number will be used to identify the patient in all future interactions with the COG. If you have problems with registration, please refer to the online help in the eRDE area of the COG website.
In order for an institution to maintain COG membership requirements, every newly diagnosed patient needs to be offered participation in ACCRN07, Protocol for the Enrollment on the Official COG Registry, The Childhood Cancer Research Network (CCRN).
A Biopathology Center (BPC) number will be assigned as part of the registration process. Each patient will be assigned only one BPC number per COG Patient ID. Each patient will be assigned only one PBC number per COG Patient ID. For additional information about the labeling of specimens, please refer to the pathology and/or Biology guidelines in this protocol.
3.1.3 Enrollment on Biology Study ANBL00B1
Enrollment on ANBL00B1 is required for all newly diagnosed patients within 21 days of diagnosis. Tissue procurement is mandatory for biology study registration. For patients with stage 4S disease who are very ill and in whom an open biopsy to obtain tissue for diagnosis and biologic studies is considered medically contraindicated, every effort should be made to obtain some tumor tissue by either fine needle aspiration of a metastatic site of disease and/or sampling of involved bone marrow, so that this tumor sample can be submitted for MYCN determination. Consent for ANBL00B1 must be obtained at the time of tissue submission and should be within one week of surgery. Needle biopsies are not sufficient for histologic classification. Investigators are strongly encouraged to obtain adequate tissue (see Sections 13.0, 14.0, and 15.0) via open biopsy techniques. For patients with Stage 4 disease who are days of age with unequivocal neuroblasts in the bone marrow, in whom a diagnostic biopsy is not obtained, a minimum of of involved bone marrow and a blood specimen must be sent to the reference lab per the requirements of ANBL00B1 to be eligible for this study. (See ANBL00B1 protocol for specifics)
3.1.4 Study Enrollment on ANBL0532
Patients may be enrolled on the study once all eligibility requirements for the study have been met. Study enrollment is accomplished by going to the Enrollment application in the eRDE system. If you have problems with enrollment, refer to online help in the Applications area of the COG website.
3.1.5 Timing
Patients must be enrolled on ANBL00B1 prior to the time of enrollment on ANBL0532. In emergency situations (or if in the opinion of the treating physician, it is in the patient's best interest) consent can be obtained and patient enrolled on ANBL00B1 and subsequently enrolled on ANBL0532 as soon as the assignment of "High-Risk" has been made in the eRDE system. When ANBL0532 enrollment is done prior to start of beginning protocol therapy, the date protocol therapy is projected to start must be no later than five (5) calendar days after enrollment. In a case of a need for emergency therapy, protocol therapy may start before enrollment on ANBL0532; however, ANBL0532 consent part 1 must be obtained prior to start of therapy AND enrollment must take place within fourteen (14) calendar days of beginning protocol therapy. Study enrollment must occur within 4 weeks of diagnosis or after only one cycle of chemotherapy on the low/intermediate risk neuroblastoma studies, or within 4 weeks of progression to stage 4 for INSS stage .
3.1.6 Bilingual Services
To allow non-English speaking patients to participate in the study, bilingual health care services will be provided in the appropriate language.
3.1.7 Randomization
Randomization will take place at completion of Induction phase of therapy via RDE. Patients will be assigned to either single myeloablative therapy or tandem myeloablative therapy. Randomization will be stratified by initial stage of disease, biologic characteristics and response to induction chemotherapy.
3.1.8 Non-Randomized Consolidation therapy:
Patients 365 to 547 days of age ( months) with Stage 4, MYCN nonamplified tumor with unfavorable histopathology or diploid DNA content or with indeterminant histology or ploidy and patients who are greater than 547 days of age with Stage 3, MYCN nonamplified tumor AND unfavorable histopathology or indeterminant histology will be nonrandomly assigned to single myeloablative transplant arm.
3.2 Patient Criteria
Important note: The eligibility criteria listed below are interpreted literally and cannot be waived (per COG policy posted ). All clinical and laboratory data required for determining eligibility of a patient enrolled on this trial must be available in the patient's medical/research record which will serve as the source document for verification at the time of audit.
3.2.1 Age
Patients must be years of age at the time of initial diagnosis.
3.2.2 Diagnosis
Patients must have a diagnosis of neuroblastoma (ICD-O morphology 9500/3) or ganglioneuroblastoma verified by histology or demonstration of clumps of tumor cells in bone marrow with elevated urinary catecholamine metabolites. Patients with the following disease stages at diagnosis are eligible, if they meet the other specified criteria. See Appendix VII.
3.2.2.1
Patients with newly diagnosed neuroblastoma with INSS Stage 4 are eligible with the following:
a. amplification (greater than four-fold increase in signals as compared to reference signals), regardless of age or additional biologic features.
b. Age > 18 months (>547 days) regardless of biologic features.
c. Age 12-18 months (365-547 days) with any of the following three unfavorable biologic features (MYCN amplification, unfavorable pathology and/or DNA index = 1) or any biologic feature that is indeterminant/unsatisfactory/unknown.
3.2.2.2
Patients with newly diagnosed neuroblastoma with INSS Stage 3 are eligible with the following:
a. amplification (greater than four-fold increase in signals as compared to reference signals), regardless of age or additional biologic features
b. Age > 18 months (> 547 days) with unfavorable pathology, regardless of status.
3.2.2.3
Patients with newly diagnosed INSS Stage 2a/2b with amplification (greater than four-fold increase in signals as compared to reference signals), regardless of age or additional biologic features.
3.2.2.4
Patients with newly diagnosed INSS Stage 4s with MYCN amplification (greater than four-fold increase in signals as compared to reference signals), regardless of additional biologic features.
3.2.2.5
Patients days initially diagnosed with: INSS stage who progressed to a stage 4 without interval chemotherapy. These patients must have been enrolled on ANBL00B1. It is to be noted that study enrollment must occur within 4 weeks of progression to Stage 4 for INSS Stage 1, 2, 4S.
3.2.3 Prior Therapy
Patients must have had no prior systemic therapy except for localized emergency radiation to sites of lifethreatening or function-threatening disease and/or no more than one cycle of chemotherapy per low or intermediate risk neuroblastoma therapy (P9641, A3961, ANBL0531) prior to determination of MYCN amplification and histology.
3.2.4 Organ Function Requirements:
3.2.4.1 Adequate renal function defined as:
Creatinine clearance or radioisotope or
Children's Oncology Group
A serum creatinine based on age/gender as follows:
Age
Maximum Serum Creatinine (mg/dL)
Male
Female
1 month to < 6 months
0.4
0.4
6 months to < 1 year
0.5
0.5
1 to < 2 years
0.6
0.6
2 to < 6 years
0.8
0.8
6 to < 10 years
1
1
10 to < 13 years
1.2
1.2
13 to < 16 years
1.5
1.4
years
1.7
1.4
The threshold creatinine values in this Table were derived from the Schwartz formula for estimating GFR (Schwartz et al. J. Peds, 106:522, 1985) utilizing child length and stature data published by the CDC.
3.2.4.2 Adequate liver function defined as:
Total bilirubin upper limit of normal (ULN) for age, and
SGOT (AST) or SGPT (ALT) < 10 x upper limit of normal (ULN) for age.
3.2.4.3 Adequate cardiac function defined as:
Shortening fraction of by echocardiogram, or
Ejection fraction of by radionuclide angiogram.
3.2.4.4 Ability to Tolerate PBSC Collection:
No known contraindication to PBSC collection. Examples of contraindications might be a weight or size less than the collecting institution finds feasible, or a physical condition that would limit the ability of the child to undergo apheresis catheter placement (if necessary) and/or the apheresis procedure.
3.2.5 Regulatory
3.2.5.1
All patients and/or their parents or legal guardians must sign a written informed consent.
3.2.5.2
All institutional, FDA, and NCI requirements for human studies must be met.
Identification of COG Approved Transplant Center: Patients entered on study must have their stem cell transplant performed at a COG approved transplant center. A COG-approved transplant center must be identified at the time of study registration. This center may be changed to another COG-approved transplant center after registration if required due to personal or financial patient issues.
3.2.6 Exclusion Criteria
3.2.6.1 Females of childbearing potential must have a negative pregnancy test. Patients of childbearing potential must agree to use an effective birth control method.
3.2.6.2 Female patients who are lactating must agree to stop breast-feeding.
3.2.6.3 Patients that are 12-18 months of age with INSS Stage 4 and all favorable biologic features (ie, non-amplified , favorable pathology, and DNA index ) are not eligible.
4.0 TREATMENT PLAN
Timing of protocol therapy administration, response assessment studies, and surgical interventions are based on schedules derived from the experimental design or on established standards of care. Minor unavoidable departures (up to 72 hours) from protocol directed therapy and/or disease evaluations (and up to 1 week for surgery) for valid clinical, patient and family logistical, or facility, procedure and/or anesthesia scheduling issues are acceptable per COG administrative Policy 5.14 (except where explicitlv prohibited within the protocol).
4.1 General Guidelines
This study will be a Phase III trial that analyses the use of intensified consolidation in high-risk neuroblastoma.
Myeloblative consolidation therapy is given at the completion of induction therapy and is categorized into Regimen A and Regimen B. Regimen A consists of one myeloablative consolidation with a CARBOplatin - etoposide - melphalan (CEM) preparative regimen. For Regimen B, patients will receive two myeloablative consolidations: Thiotepa - cyclophosphamide (TC) preparative regimen followed by CEM preparative regimen.
Most patients will be randomized into either Regimen A or Regimen B. Randomization will be stratified by stage, MYCN status and response to induction therapy.
The following patients will be nonrandomly assigned to single myeloablative transplant (Regimen A):
Patients who are 365 to 547 days of age ( 12 - 18 months) with Stage 4, MYCN nonamplified tumor with any of the following potentially unfavorable biologic features:
unfavorable histopathology
diploid DNA content
indeterminant histopathology
indeterminate ploidy
Patients who are greater than 547 days of age with Stage 3, MYCN nonamplified tumor with either of the following:
unfavorable histopathology
indeterminant histopathology
Patients who develop progressive disease (PD) at the end of induction, those who are unable to obtain adequate PBSC for transplant or who have GFR will be taken off protocol therapy. Refer to protocol section 8.1.1 for complete definition of off protocol therapy criteria.
4.1.1 Central Line
All patients will have a double lumen central venous line placed prior to beginning induction chemotherapy.
4.1.2
Chemotherapy doses for all drugs EXCEPT topotecan will be adjusted for patients who weigh ; these patients will have chemotherapy dosed per kg rather than per body surface area (BSA). Dosing will be adjusted only during cycles when patient's weight is . Topotecan dosing will be based on BSA regardless of age or weight.
4.1.3 Induction Therapy
Patients will receive 6 cycles of induction chemotherapy. Chemotherapy cycles (except Cycle 1) may begin when the ANC and platelets after post-chemotherapy nadir. There are no hematologic criteria to begin Cycle 1.
Children's Oncology Group
Patients who received one cycle of chemotherapy per low or intermediate risk neuroblastoma therapy (P9641, A3961, ANBL0531) prior to determination of MYCN amplification and histology will receive all 6 cycles of ANBL0532 induction therapy.
4.1.3.1
Patients will undergo surgical resection after Cycle 5 (or Cycle 6, if medically necessary). Begin next chemotherapy cycle as soon as possible following surgery. Therapy should not be delayed by more than one week unless complications arise. If surgery cannot be scheduled after Cycle 5, it should occur after Cycle 6.
4.1.3.2
Primary and metastatic tumor response will be assessed after 2 cycles and at completion of induction chemotherapy. Primary site tumor assessment prior to surgical resection (Cycle 5 or Cycle 6, if medically necessary) is mandatory as the pre-surgical primary tumor volume will be used to establish radiation fields.
4.1.3.3
Patients with progressive disease (PD) at the end of induction evaluation will be taken off protocol therapy.
4.1.3.4 Peripheral Blood Stem Cell Harvest (PBSC) (See Section 19.0 for complete details)
Patients will undergo PBSC harvest after 2 cycles REGARDLESS of persistent bone marrow metastatic disease. There will not be any no ex vivo manipulation of PBSC prior to cryopreservation as results from the randomized question in A3973 did not demonstrate a benefit in survival following ex vivo purging of PBSC product.
PBSC collection will be performed prior to randomization. Therefore, all patients must have a PBSC collection that is sufficient for tandem transplant with a goal for collection of divided into 3 separate aliquots. A minimum number of frozen PBSC of CD34 cells divided into 2 aliquots is required for all patients ( CD34 cells for each transplant) to remain eligible for transplant while an additional aliquot of CD34 cells is strongly recommended to be stored for back-up.
4.2 Induction Therapy Administration
For patients , chemotherapy doses will be calculated by body surface area (BSA).
For patients , chemotherapy doses for all drugs EXCEPT topotecan will be dosed per kg rather than per BSA.
Topotecan dosing will be based on BSA regardless of age or weight.
Note specific dosing instructions for vinCRIStine in Cycles 4 and 6 of induction.
Myeloid Growth Factors: During induction cycles 1 and 3-6 (all induction cycles except cycle 2 or other cycles in which PBSC collections are planned), cytokine support need not be limited to G-CSF i.e: other growth factors are permitted according to an institution's standard guidelines. Choice of myeloid growth factor must be recorded appropriately in the therapy delivery maps. Myeloid growth factors (including GCSF) should be administered hours after the last dose of chemotherapy. Daily myeloid growth factor therapy should be continued until the ANC . Discontinue daily myeloid growth factor support a minimum of 24 hours prior to administration of the next chemotherapy cycle. See Appendix II for supportive care guidelines.
Institutions are permitted to use their standard guidelines for hydration/monitoring parameters pertaining to chemotherapy agents utilized in this protocol. In the absence of institutional standards, suggested guidelines are included in the therapy delivery maps that follow.
4.2.1 Induction Therapy Cycle 1
COG #
Induction Therapy Cycle 1
Induction Therapy Cycle 1
DOB
No hematologic criteria to begin Cycle 1. This course lasts 21 days. This Therapy Delivery Map is on one page.
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Cyclophosph amide (CPM)
IV over 30-60 minutes
dose (or if , ) once daily x 5 doses.
Days 1-5
Suggest hydrating at with fluid containing at least for 2 hours prior to and 2 hours after each dose.
Topotecan (TOPO)
IV over 30 minutes
dose (all patients) once daily x 5 doses.
Days 1-5
i. Peripheral blood for immune assessments
i. Peripheral blood for immune assessments (Section 15.8) (Section 15.8)
Myeloid growth factor Adminsitration: Begin 24 - 48 hours after completion of chemotherapy.If given daily, continue until post-nadir ANC . See sections 4.2 and Appendix II for specific directions.
a. Physical, Ht, Wt
b. CBC with diff and platelets
c. Electrolytes, BUN,
d. ALT, AST, Bilirubin, Urinalysis, Albumin
e. See Section 7.1.1 for pre-treatment evaluations.
f. Pharmacogenomics (Section 15.2)
g. BM ICC and PCR (Section 15.4)
h. Peripheral blood PCR (Section 15.6)
j. Topotecan Pharmacokinetics (Section 15.3)
Refer to Protocol Section 7.0 for complete list of observations and Section 15.0 for sample acquisition or shipment guidelines
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Therapy Delivery Map
Cycle 1
Ht
Wt
Date Due
Date Given
Day
CPM mg
TOPO mg
Myeloid growth factor used:
mcg
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
a,b,c,d,e,f,g,h,i
1
CPM mg
TOPO_ mg
j
2
CPM mg
TOPO mg
3
CPM mg
TOPO mg
4
CPM mg
TOPO mg
5
CPM mg
TOPO mg
6
___mcg
b (obtain twice weekly while on myeloid growth factor)
Date of last dose:
21
End of cycle
22
Begin next cycle on Day 22
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
NOTE: For eRDEs reporting: use Day 22 (or treatment start date for next cycle if > Day 22) as the end date for the current cycle.
Page 1 of 1
4.2.2 Induction Therapy Cycle 2
COG #
Induction Therapy Cycle 2
Patients should begin therapy on day 22 of the prior cycle or as soon thereafter as and platelets . This course lasts 21 days. This Therapy Delivery Map is on one page.
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Cyclophosphamide (CPM)
IV over 30 60 minutes
dose (or if mg/kg/dose) once daily x 5 doses.
Days 1-5
Suggest hydrating at with fluid containing at least for 2 hours prior to and 2 hours after each dose.
Topotecan (TOPO)
IV over 30 minutes
dose (all patients) once daily x 5 doses.
Days 1-5
Topotecan dosing will be based on BSA regardless of age or weight.
Filgrastim (GCSF)
SubQ or IV (SubQ preferred)
5 micrograms/kg beginning 24 hours after completion of chemotherapy and continuing once daily until post-nadir ANC . Once postnadir ANC , increase dose to 10 micrograms/kg and continue once daily until PBSC harvest is complete.
Day 6
Discontinue at least 24 hours prior to next chemotherapy cycle.
a. Physical, Ht, Wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr, Ca, Phos, Mg
d. ALT, AST, Bilirubin, Urinalysis e. Topotecan Pharmacokinetics (Section 15.3)
f. PBSC PCR (Section 15.5)
g. Bilateral BM Asp/Bx
h. BM PCR (Section 15.4)
i. Tumor Imaging, Bone scan, MIBG (Section 7.1.1)
j. MRI Spine & Neuro Eval (tumors with intraspinal extension only)
k. VMA, HVA (if elevated at diagnosis)
Refer to Protocol Section 7.0 for complete list of observations and Section 15.0 for sample acquisition or shipment guidelines
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Therapy Delivery Map Cycle 2
Ht
cm
Wt
kg BSA
Date Due
Date Given
Day
CPM mg
TOPO mg
GCSF mcg
Studies
Comments (Include any held doses, or dose modifications)
a,b,c,d,
1
CPM
mg
TOPO
e
2
CPM
mg
TOPO
3
CPM
mg
TOPO
4
CPM
mg
TOPO
5
CPM
mg
TOPO
6
GCSF
b (obtain twice weekly while on GCSF)
Date of last dose:
14
Collect PBSCs (See Protocol Section 19.0)
f
21
End of cycle
g,h,i,j,k
22
Begin next cycle on Day 22
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
NOTE: For eRDEs reporting: use Day 22 (or treatment start date for next cycle if > Day 22) as the end date for the current cycle.
Page 1 of 1
Children's Oncology Group
4.2.3 Induction Therapy Cycle 3
COG #
Induction Therapy Cycle 3
Induction Therapy Cycle 3
Patient name or initials
DOB
Patients should begin each cycle of induction therapy on Day 22 of the prior cycle or as soon thereafter as ANC and platelets .
This course lasts 21 days. This Therapy Delivery Map is on one page.
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
CISplatin (CDDP)
IV over 1 hour
dose (or if dose once daily x 4 doses.
Days 1-4
Suggest hydrating at 3000 day using fluid containing at least 0.45% NaCl. Suggest achieving urine specific gravity 1.010 prior to start of CISplatin. Hydration fluids may contain supplemental magnesium, calcium and potassium to decrease electrolyte losses associated with cisplatin. Administration of mannitol per institutional guidelines is recommended.
a. Physical, Ht, Wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr, Ca, Phos, Mg
d. ALT, AST, Bilirubin, Urinalysis
Refer to Protocol Section 7.0 for complete list of observations
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Etoposide (ETOP)
IV over 1 hour
dose (or if dose ) once daily x 3 doses.
Days 1-3
Myeloid growth factor Adminsitration: Begin 24 - 48 hours after completion of chemotherapy.If given daily, continue until post-nadir ANC . See sections 4.2 and Appendix II for specific directions.
Therapy Delivery Map Cycle 3
Ht cm
Wt kg
BSA
Date Due
Date Given
Day
CDDP mg
ETOP mg
Myeloid Growth Factor: Dose: mcg
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
a,b,c,d
1
CDDP mg
ETOP mg
2
CDDP mg
ETOP mg
c
3
CDDP mg
ETOP mg
c c
4
CDDP mg
5
mcg
b (obtain twice weekly while on myeloid growth factor)
Date of last dose:
21
End of cycle
Begin next cycle on Day 22
Page 1of 1
4.2.4 Induction Therapy Cycle 4
Induction Therapy Cycle 4
Note specific dosing instructions for vinCRIStine.
Patient name or initials
DOB
Patients should begin each cycle of induction therapy on day 22 of the prior cycle or as soon thereafter as and platelets . This cycle lasts 21 days. This Therapy Delivery Map is on one page.
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Cyclophosphamide (CPM)
IV over 6 hours
dose (or if dose ) once daily doses.
Days 1-2
Suggest hydrating at 3000 day using fluid containing at least NaCl . Suggest achieving urine specific gravity 1.010 prior to start of cyclophosphamide.
a. Physical, Ht, Wt
b. CBC with diff and platelets
c. Electrolytes, BUN,
d. ALT, AST, Bilirubin, Urinalysis
Refer to Protocol Section 7.0 for complete list of observations.
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Mesna (MESNA)
IV over 15 minutes
dose (or if dose ) immediately prior to each cyclophosphamide and again at 4 and 8 hours after each cyclophosphamide infusion
Days 1-2
DOXOrubicin (DOXO)
IV over 24 hours
dose (or if dose ) once daily doses.
Days 1-3
VinCRIStine (VCR)
IV push over 1 minute (or) infusion via minibag as per institutional policy
Patients < months of age: 0.017 dose once daily x 3 doses
Patients months and : dose or dose (whichever is LOWER for all patients > 12 kg ) once daily doses
Patients months and : dose once daily x 3 doses
Days 1-3
NOTE: total dose may not exceed 2 mg in 72 hours or day for any patient
VinCRIStine should be administered prior to start of DOXOrubicin infusion and then daily for a total of 3 doses.
Myeloid growth factor Adminsitration: Begin 24-48 hours after completion of chemotherapy. If given daily, continue until post-nadir ANC . See sections Section 4.2 and Appendix II for specific directions.
Therapy Delivery Map Cycle 4
Ht
cm
Wt kg
BSA
Date Due
Date Given
Day
CPM
Mesna
Doxo
VCR mg
Myeloid Growth
Factor:
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
a, b, c, d
1
CPM mg
Mesna mg
Doxo_mg
VCR_ mg
Mesna_mg
Mesna_mg
2
CPM mg
Mesna_mg
Doxo_mg
VCR_ mg
Mesna_ mg
Mesna_mg
3
Doxo_ mg
VCR_ mg
4
5
__ mcg
b (obtain twice weekly while on myeloid growth factor)
Date of last dose:
21
22
Begin next cycle on day 22
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
NOTE: For eRDEs reporting: use Day 22 (or treatment start date for next cycle if > Day 22) as the end date for the current cycle.
4.2.5 Induction Therapy Cycle 5
COG #
Induction Therapy Cycle 5
Patients will undergo surgical resection after Cycle 5 (or Cycle 6, if medically necessary). Begin next chemotherapy cycle as soon as possible following surgery. Therapy should not be delayed by more than one week unless complications arise. If surgery cannot be scheduled after Cycle 5, it should occur after Cycle 6.
Patient name or initials
DOB
Patients should begin each cycle of induction therapy on Day 22 of the prior cycle or as soon thereafter as ANC and platelets . This course lasts 21 days. This Therapy Delivery Map is on one page.
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
CISplatin (CDDP)
IV over 1 hour
dose (or if mg/kg/dose) once daily x 4 doses.
Days 1-4
Suggest hydrating at 3000 day using fluid containing at least 0.45% NaCl. Suggest achieving urine specific gravity prior to start of CISplatin. Hydration fluids may contain supplemental magnesium, calcium and potassium to decrease electrolyte losses associated with CISplatin. Administration of mannitol per institutional guidelines is recommended.
a. Physical, Ht, Wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr, Ca , Phos, Mg
d. ALT, AST, Bilirubin, Urinalysis
e. Primary tumor imaging
Refer to Protocol Section 7.0 for complete list of observations.
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Etoposide (ETOP)
IV over 1 hour
dose (or if , dose ) once daily x 3 doses.
Days 1-3
Myeloid growth factor Administration: Begin 24-48 hours after completion of chemotherapy. If given daily, continue until post-nadir ANC . See sections 4.2 and Appendix II for specific directions.
Therapy Delivery Map
Cycle 5
Ht
cm Wt
kg BSA
Studies
Date Due
Date Given
Day
CDDP mg
ETOP mg
Myeloid Growth Factor : mcg
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
a,b,c,d
1
CDDP _ mg
ETOP_ mg
c
2
CDDP mg
ETOP _ mg
3
CDDP mg
ETOP_ mg
c
4
CDDP
c
5
mcg
b (obtain twice weekly while on myeloid growth factor)
Date of last dose:
21
Surgery following Cycle 5
Begin next cycle on Day 22
e(prior to surgery)
@ - Please note that this test is only recommended to be done on Days 1, 2, 3 and 4
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
NOTE: For eRDEs reporting: use Day 22 (or treatment start date for next cycle if Day 22 ) as the end date for the current cycle.
Page 1
4.2.6 Induction Therapy Cycle 6
COG #
Induction Therapy Cycle 6
Note specific dosing instructions for vinCRIStine.
Patients should begin each cycle of induction therapy on Day 22 of the prior cycle or as soon thereafter as ANC and platelets . This cycle lasts 21 days and this Therapy Delivery Map is on one page.
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Cyclophosphamide (CPM)
IV over 6 hours
dose (or if dose ) once daily doses.
Days 1-2
Hydrate at 3000 day using fluid containing at least . Achieve urine specific gravity prior to start of cyclophosphamide.
a. Physical, Ht, Wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr, Ca, Phos, Mg
d. ALT, AST, Bilirubin, Urinalysis, Albumin
e. ECG, MUGA/EHCO, CrCl or GFR, Audiogram/BAER (see Section 7.1.1)
f. Bilateral BM ASP/Bx
g. BM ICC and PCR (Section 15.4)
h. Peripheral Blood PCR (Section
Mesna (MESNA)
IV over 15 minutes
dose (or if mg/kg/dose) immediately prior to each cyclophosphamide and again at 4 and 8 hours after each cyclophosphamide infusion.
Days 1-2
DOXOrubicin (DOXO)
IV over 24 hours
dose (or if ) once daily doses.
Days 1-3
15.6)
i. Tumor Imaging, Bone scan,
Patients < months of age: 0.017 mg/kg/dose once daily x 3 doses
Patients months and : dose or 0.022 dose (whichever is LOWER for all patients ) once daily x 3 doses
Patients months and : dose once daily doses
Days 1-3
NOTE: Total dose may not exceed 2 mg in 72 hours or 0.67 mg/day for any patient
VinCRIStine should be administered prior to start of DOXOrubicin infusion and then daily for a total of 3 doses.
MIBG (Section 7.1.1)
j. MRI Spine & Neuro Eval (tumors with intraspinal extension only) k. VMA, HVA (if elevated at diagnosis)
Refer to Protocol Section 7.0 for complete list of observations and Section 15.0 for sample acquisition or shipment guidelines
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Myeloid growth factor Administration: Begin 24-48 hours after completion of chemotherapy. If given daily, continue until post-nadir ANC . See sections 4.2 and Appendix II for specific directions.
Cycle 6
Wt
kg BSA
Date Due
Date Given
Day
CPM __mg
Mesna mg
Doxo mg
VCR mg
Myeloid Growth Factor : mcg
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
1
CPM_ mg
Mesna__mg
Doxo_mg
VCR_mg
Mesna_ mg
Mesna_ mg
2
CPM mg
Mesna__mg
Doxo ... mg
VCR_ mg
Mesna_mg
Mesna_mg
3
Doxo ...mg
VCR_mg
4
5
__mcg
b (obtain twice weekly while on myeloid growth factor)
Date of last dose:
21
Proceed to randomization once staging and organ function evaluations are complete Proceed to Consolidation Therapy (see Section 4.4)
a,b,c,d,e,f,g,h,i,j,k
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
NOTE: For eRDEs reporting: use Day 22 (or treatment start date for next cycle if > Day 22) as the end date for the current cycle.
Page 1 of 1
ANBL0532
4.3 Local Control - Surgery
Surgical resection of soft tissue disease will occur during induction therapy. Refer to section 13.0 for complete details.
4.3.1 Timing of Surgery
Second look surgery
Residual persistent mass after initial chemotherapy is common, and in previous analyses is not correlated with patient outcome. All patients will undergo attempt at complete surgical resection of primary tumor following Cycle 5 (or Cycle 6, if medically necessary) of induction chemotherapy.
4.4 Consolidation Therapy
Eligibility to proceed to Consolidation Therapy/Transplant: Only patients who have a minimum number of frozen PBSC of CD cells (minimum of CD cells transplant), who have adequate organ function, who do not have progressive disease at end of induction AND who have consented to Consent Part 2 are eligible for transplantation.
Eligibility for randomization: Patients must first be eligible to proceed to transplant in order to be eligible for randomization. Patients will be randomly assigned to either single myeloablative therapy (Regimen A) with a CARBOplatin, etoposide, melphalan (CEM) preparative regimen and autologous PBSC rescue (representing the standard arm of the study), to the experimental arm of tandem myeloablative therapy (Regimen B) with the regimen used in the COG ANBL00P1 (thiotepa and cyclophosphamide followed by CEM).
The following patients will be nonrandomly assigned to single myeloablative transplant (Regimen A):
Patients who are 365 to 547 days of age ( months) with Stage 4, MYCN nonamplified tumor with any of the following potentially unfavorable biologic features:
unfavorable histopathology
diploid DNA content
indeterminant histopathology
indeterminate ploidy
Patients who are greater than 547 days of age with Stage 3, MYCN nonamplified tumor with either of the following:
unfavorable histopathology
indeterminant histopathology
Patients should begin consolidation chemotherapy no later than 8 weeks after the start of Induction Cycle #6. (It is strongly recommended to begin consolidation within 4-6 weeks after starting Induction Cycle #6.) Patients who are delayed beyond 8 weeks, but who still meet organ function criteria should remain on protocol therapy and proceed to transplant. Delay in starting transplant will be considered a protocol deviation.
Patients with progressive disease at end of induction therapy are NOT eligible to continue on protocol therapy. Patients with mixed response or stable disease evaluation may remain on protocol therapy as per investigator preference. However, such patients are encouraged to come off protocol therapy for entry onto available COG Phase I and Phase II studies.
No restaging will be performed between Hematopoietic Stem Cell Transplants (HSCT) or prior to radiation therapy unless clinically indicated. Patients randomized to Regimen B will proceed to HSCT #2 (CEM) no less than 6 and prior to 10 weeks from day 0 of HSCT #1, providing they have recovered from acute toxicities of HSCT#1 and meet organ eligibility criteria for HSCT #2.
4.5 Consolidation Therapy Regimen A: Single HSCT (CEM)
COG #
Regimen A: single myeloablative therapy with a CARBOplatin, etoposide, melphalan (CEM) preparative regimen and autologous PBSC rescue (representing the standard arm of the study).
Patients should begin consolidation therapy within 8 weeks of beginning Cycle 6 of induction. This Therapy Delivery Map is on FOUR pages. This cycle lasts 36 days.
Criteria to Start Consolidation Regimen A:
No evidence of disease progression: defined as increase in tumor size of or new lesions.
Recovery from last induction course of chemotherapy.
No uncontrolled infection.
Minimum frozen PBSC of cells as 2 aliquots; cells for transplant are mandatory and cells for back-up are strongly recommended.
AST upper normal
Shortening fraction , or ejection fraction , no clinical congestive heart failure.
Creatinine clearance or (If a creatinine clearance is performed at end induction and the result is , a GFR must then be performed using a nuclear blood sampling method or iothalamate clearance method. Camera method is NOT allowed as measure of GFR prior to or during Consolidation therapy for patients with GFR or creatinine clearance of .)
Refer to Protocol Section 7.0 for complete list of observations and Section 15.0 for sample acquisition or shipment guidelines
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
*Calvert Formula for CARBOplatin dosing:
Patients with GFR who are will be dosed according to the formula below to achieve an area under the concentration versus time curve (AUC) of 4.1 per dose. GFR will be calculated using a blood sampling method. Daily dose should not exceed .
Example: Patient's GFR is , they weigh 14.6 kg and their BSA is :
NOTE: if lab reports raw GFR in instead of corrected GFR in , convert Raw GFR to corrected GFR as follows:
Divide Raw GFR in by the patient's surface area in , then multiply result by .
Example: Raw GFR is divided by then multiplied by
Page 2 of 4
Consolidation Regimen A: Single HSCT (CEM): Patients with altered renal function who are PATIENTS WITH GFR AND ARE
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Melphalan (MEL)
IV over 15-30 minutes
dose once daily x 3 doses.
Days -7, -6 and -5
Must be infused within 1 hour of preparation.
Etoposide (ETOP)
IV over 24 hours
dose once daily x 4 doses.
Days -7, -6, -5 and -4
CARBOplatin (CARB)
IV over 24 hours
See Calvert Formula* below for dose calculation; give dose once daily x 4 doses. Also calculate dose as 10 and give LOWEST calculated dose as described below
Days -7, -6, -5 and -4
*See sample Calvert dose calculation below.
Filgrastim (GCSF)
SubQ (OR) IV (SubQ preferred)
5 micrograms/kg once daily and continuing once daily until post-nadir ANC for 3 consecutive days.
Day 0 until ANC recovery
a. Physical exam, ht, wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr
d.
e. ALT, AST, Bilirubin, Albumin
f. Triglycerides
g. ECG, MUGA/ECHO, GFR, urinalysis
h. Bilateral BM aspirate/bx., Audiogram/BAER (Section 7.1.2)
n. Peripheral Blood Immune assessments (Section 15.8)
o. Peripheral blood for Immune recovery (Section 7.1.2)
Refer to Protocol Section 7.0 for complete list of observations and Section 15.0 for sample acquisition or shipment guidelines
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
*Calvert Formula for CARBOplatin dosing:
Patients with GFR who are will be dosed according to the formula below to achieve an area under the concentration versus time curve (AUC) of 4.1 per dose. GFR will be calculated using a blood sampling method. Daily dose should not exceed . Calculate dose by Calvert formula below and also calculate dose by weight ( ). Give LOWER of the two calculated doses.
Total Dose day Raw GFR weight in kg
Example: Patient's GFR is , they weigh 10.2 kg and their BSA is :
To convert corrected GFR reported as to Raw GFR reported as , multiply corrected GFR by the patient's BSA, then divide by : then divided by
Total dose day
Page 3 of 4
Consolidation Regimen A: Single HSCT (CEM)
For tests required prior to consolidation, please refer to Section 7.1.2
Therapy Delivery Map
cm
Wt
kg
Date Due
Date Given
Day
MEL mg
ETOP mg
CARB mg
GCSF mcg
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
-7
MEL
ETOP
mg
CARB
mg
a,b,c,d,e
-6
MEL
ETOP
mg
CARB
mg
b
-5
MEL
ETOP
mg
CARB
mg
b,c
-4
ETOP
mg
CARB
mg
b
-3
b
-2
b,c
-1
b
0
GCSF___mcg
mcg
(daily until neutrophil engraftment), c (every other day if stable), d, e (twice weekly if stable)
Date of last dose:
28
Begin radiation therapy no sooner than 28 days post transplant, and
o (1 month post HSCT), b (weekly through radiation therapy),
End of radiation therapy:a,b,c,d,e,f,g,h,i,j,k,l,m,n,o
Please note that tests from Day -6 to Day -1 are recommended only.
Children's Oncology Group
4.6 Consolidation Therapy Regimen B: Tandem HSCT #1 (Thiotepa and Cyclophosphamide)
COG #
Regimen B: Experimental arm of tandem myeloablative therapy (Regimen B) with thiotepa and cyclophosphamide regimen followed by CEM.
Patient name or initials
DOB
Patients should begin consolidation therapy within 8 weeks of beginning Cycle 6 of induction.
This Therapy Delivery Map is on two pages. This cycle lasts 50 days
Criteria to Start Consolidation Regimen B: Tandem HSCT #1 (Thiotepa and Cyclophosphamide)
No evidence of disease progression: defined as increase in tumor size of or new lesions.
Recovery from last induction course of chemotherapy. 3.No uncontrolled infection.
Minimum frozen PBSC of cells as 2 aliquots; i.e. cells for each transplant are mandatory. A third aliquot of cells is strongly recommended for back-up.
5.AST upper normal
Shortening fraction , or ejection fraction , no clinical congestive heart failure.
Creatinine clearance or GFR (If a creatinine clearance is performed at end of induction and the result is , a GFR must be performed using a nuclear blood sampling method or iothalamate clearance method. Camera method is NOT allowed as measure of GFR prior to or during Consolidation therapy for patients with GFR or creatinine clearance of .)
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Thiotepa (TEPA)
IV over 2 hours
dose (or if dose ) once daily x 3 doses.
Days -7, -6 and -5
Thiotepa can cause significant skin toxicity with sloughing of skin. Bathe patient frequently in water only. Avoid large occlusive dressings, use of any skin creams and remove adhesive residue from prior dressings and leads.
a. Physical exam, ht, wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr,
d.
e. ALT, AST, bilirubin, albumin
Refer to Protocol Section 7.0 for complete list of observations.
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Cyclophosphamide (CPM)
IV over 1 hour
dose (or if dose ) once daily x 4 doses.
Days -5, -4, -3, and -2
Suggest hydrating at 3000 day using fluid containing at least 0.45% NaCl. Suggest achieving urine specific gravity 1.010 prior to start of cyclophosphamide.
Mesna
IV over 15 minutes
dose (or if dose ) immediately prior to each cyclophosphamide dose and then 4 hours and 8 hours after each cyclophosphamide dose
Days -5, -4, -3, and -2
Filgrastim (GCSF)
SubQ (or) IV (SubQ preferred)
5 micrograms/kg once daily and continuing once daily until post-nadir ANC > for 3 consecutive days.
Day 0 until ANC recovery
Page 1 of 2
Consolidation Therapy Regimen B: Tandem HSCT #1 (Thiotepa and Cyclophosphamide)
For tests required prior to consolidation, please refer to Section 7.1.2
Therapy Delivery Map
Ht__cm Wt__kg
Date Due
Date Given
Day
TEPA mg
CPM mg
Mesna mg
GCSF mcg
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
-7
TEPA mg
a,b,c,d,e
-6
TEPA mg
b,c
-5
TEPA_mg
CPM_ mg
Mesna_mg
b
Mesna_ mg
Mesna_mg
-4
CPM_ mg
Mesna__mg
b,c
Mesna_ mg
Mesna_mg
-3
CPM_ mg
Mesna__mg
b
Mesna_ mg
Mesna__mg
-2
CPM mg
Mesna_mg
b,c
Mesna_mg
Mesna__mg
-1
0
PBSC infusion (HSCT #1)
GCSF___mcg
a, b (daily until neutrophil engraftment), c (every other day if stable), d, e (twice weekly if stable)
Date of last dose:
42
Proceed to HSCT #2 (CEM) no less than 6 weeks and prior to 10 weeks from day 0 of HSCT #1
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
Please note that tests from Day -6 to Day -1 are recommended only.
Page 2 of 2
ANBL0532
4.6.1 Consolidation Regimen B: HSCT #2 (CEM)
COG #
Regimen B: Experimental arm of tandem myeloablative therapy (Regimen B) with the thiotepa and cyclophosphamide regimen followed by CEM.
Patient name or initials
DOB
Criteria to Start Consolidation Regimen B HSCT#2:
No restaging will be performed between transplants or prior to radiation therapy unless clinically indicated. Patients randomized to Regimen B will proceed to HSCT #2 (CEM) no less than 6 weeks and prior to 10 weeks from Day 0 of HSCT #1, providing they have recovered from acute toxicities of HSCT#1 and meet organ eligibility criteria for HSCT #2.
All patients must have repeat GFR after recovery from HSCT #1 and prior to HSCT #2. If a creatinine clearance is performed and the result is , a GFR must be performed using a nuclear blood sampling method or iothalamate clearance method. Camera method is NOT allowed as measure of GFR prior to or during Consolidation therapy if GFR/creatinine clearance is . Patient is ineligible to receive HSCT #2 if CrCl or GFR is . If the GFR is , then modified dosing is used as noted below. It is recommended that the Study Chair be notified of patients with low glomerular filtration rates.
Resolution of acute hepatic, pulmonary or cardiac toxicities developed during HSCT #1.
AST upper normal
Shortening fraction , or ejection fraction , no clinical congestive heart failure.
No uncontrolled infection.
No moderate or severe sinusoidal obstruction syndrome (SOS) (formerly veno-occlusive disease (VOD) ) during HDC/SCR #1
Definition of sinusoidal obstruction syndrome (SOS)
SOS (formerly known as VOD) is a syndrome of hepatic dysfunction, which develops by Day+21 post stem cell transplant, and is characterized by hyperbilirubinemia , with at least two of the following three findings: ascites, hepatomegaly, which is usually painful, and weight gain over baseline. Patients with SOS during SCT #1, with hepatic function meeting eligibility for SCT#2 and NOT meeting definitions for moderate or severe SOS (see below), may proceed to SCT#2.
Definition of severe SOS:
Severe SOS is defined as a SOS episode accompanied by specific organ failure (hepatic encephalopathy (CTC Grade 4 liver dysfunction/failure, clinical will be referred as "Hepatic failure" per CTCAE v.4.0 followed starting July , 2011), continuous oxygen requirement (CTC Grade 3 hypoxia), serum creatinine times the upper limit of normal (CTC Grade 3 creatinine), requirement for ventilatory support or dialysis not clearly attributable to another cause. Patients with a history of severe SOS, even if resolved, should not undergo SCT#2.
Definition of moderate SOS:
Moderate SOS is defined as a SOS episode where the peak total bilirubin was or greater (personal communication, Paul Richardson). Patients with a history of moderate SOS, even if resolved, should not undergo SCT#2.
7. Minimum frozen PBSC of cells is mandatory to proceed with second transplant.
8. No clinical evidence of disease progression defined as increase in tumor size by or new lesions; restaging studies between HSCT courses only as clinically indicated.
9. ANC recovery to after HSCT #1
10. Platelet count or and responsive to platelet transfusion
This Therapy Delivery Map is on FIVE pages. This Cycle lasts 36 days.
Page 1 of 5
Consolidation Regimen B: HSCT #2 (CEM)
PATIENTS WITH CREATININE CLEARANCE OR GFR
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Melphalan (MEL)
IV over 15-30 minutes
dose (or if kg, dose) once daily x 3 doses.
Days -7, 6 and -5
Must be infused within 1 hour of preparation.
Etoposide (ETOP)
IV over 24 hours
dose (or if dose ) once daily x 4 doses.
Days -7, 6, -5 and 4
CARBOplatin (CARB)
IV over 24 hours
dose (or if dose ) once daily x 4 doses. .
Days -7, 6, -5 and 4
Filgrastim (GCSF)
SubQ (OR) IV (SubQ preferred)
5 micrograms/kg once daily and continuing once daily until post-nadir ANC for 3 consecutive days.
Day 0 until ANC recovery
a. Physical exam, ht, wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr
d.
e. ALT, AST, Bilirubin, Albumin
f. Triglycerides
g. ECG, MUGA/ECHO, CrCl or GFR, urinalysis
h. Bilateral BM aspirate/bx., Audiogram/BAER (Section 7.1.2)
n. Peripheral Blood Immune assessments (Section 15.8)
o. Peripheral blood for Immune recovery (Section 7.1.2)
Refer to Protocol Section 7.0 for complete list of observations and Section 15.0 for sample acquisition or shipment guidelines
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
*Calvert Formula for CARBOplatin dosing:
Patients with GFR who are will be dosed according to the formula below to achieve an area under the concentration versus time curve (AUC)
of 4.1 per dose. GFR will be calculated using a blood sampling method. Daily dose should not exceed 300 .
Total Dose
Example: Patient's GFR is , they weigh 14.6 kg and their BSA is :
Total dose day 1.73
NOTE: if lab reports raw GFR in instead of corrected GFR in , convert Raw GFR to corrected GFR as follows:
Divide Raw GFR in by the patient's surface area in , then multiply result by .
Example: Raw GFR is : divided by then multiplied by
Consolidation Regimen B: HSCT #2 (CEM): Patients with altered renal function who are
PATIENTS WITH GFR and AND ARE
DRUG
ROUTE
DOSAGE
DAYS
IMPORTANT NOTES
OBSERVATIONS
Melphalan (MEL)
IV over 15-30 minutes
dose once daily x 3 doses. .
Days -7, -6 and -5
Must be infused within 1 hour of preparation.
Etoposide (ETOP)
IV over 24 hours
dose once daily x 4 doses.
Days -7, -6, -5 and -4
CARBOplatin (CARB)
IV over 24 hours
See Calvert Formula* below for dose calculation; give dose once daily x 4 doses. . Also calculate dose as 10 and give LOWEST calculated dose as described below
Days -7, -6, -5 and -4
*See sample Calvert dose calculation below.
Filgrastim (GCSF)
SubQ (OR) IV (SubQ preferred)
5 micrograms/kg once daily and continuing once daily until post-nadir ANC for 3 consecutive days.
Day 0 until ANC recovery
a. Physical exam, ht, wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr
d.
e. ALT, AST, Bilirubin, Albumin
f. Triglycerides
g. ECG, MUGA/ECHO, CrCl or GFR, urinalysis
h. Bilateral BM aspirate/bx., Audiogram/BAER (Section 7.1.2)
n. Peripheral Blood Immune assessments (Section 15.8)
o. Peripheral blood for Immune recovery (Section 7.1.2)
Refer to Protocol Section 7.0 for complete list of observations and Section 15.0 for sample acquisition or shipment guidelines
OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
*Calvert Formula for CARBOplatin dosing:
Patients with GFR who are will be dosed according to the formula below to achieve an area under the concentration versus time curve (AUC) of 4.1 per dose. GFR will be calculated using a blood sampling method. Daily dose should not exceed . Calculate dose by Calvert formula below and also calculate dose by weight ( ). Give LOWER of the two calculated doses.
Total Dose day Raw GFR weight in kg
Example: Patient's GFR is , they weigh 10.2 kg and their BSA is :
To convert corrected GFR reported as to Raw GFR reported as , multiply corrected GFR by the patient's BSA, then divide by :
Page 4 of 5
Consolidation Regimen B: HSCT #2 (CEM)
For tests required prior to consolidation, please refer to Section 7.1.2
Therapy Delivery Map
Ht
cm
Wt
kg
Date Due
Date Given
Day
MEL mg
ETOP mg
CARB mg
GCSF mcg
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
-7
MEL mg
ETOP_ mg
CARB mg
a,b,c,d, e
-6
MEL_ mg
ETOP_ mg
CARB_mg
b,c
-5
MEL mg
ETOP mg
CARB mg
b,
-4
ETOP_ mg
CARB_mg
b,c
-3
b
-2
b,c
-1
b
0
PBSC infusion (HSCT #2)
GCSF___ mcg
a, b (daily until neutrophil engraftment), c (every other day if stable), d, e (twice weekly if stable), g
Date of last dose:
28
Begin radiation therapy no sooner than 28 days post transplant, and recommended to begin by 42 days post transplant (see Protocol section 17.0).
o (1 month post HSCT), b(weekly through radiation therapy), End of radiation therapy:a,b,c,d,e, f,g,h,i,j,k,l,m,n,o
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
Please note that tests from Day to Day are recommended only.
4.7 Radiation therapy
After completion of stem cell transplantation and recovery from acute toxicity, patients will receive radiation therapy to the primary site of disease as well as to MIBG-avid sites that were documented at pre-transplant (end-induction) evaluation. Patients who have a complete surgical resection of the primary tumor will receive 21.6 Gy external beam radiation therapy (EBRT) to the post-induction chemotherapy, pre-operative primary tumor volume while those who have an incomplete surgical resection of the primary tumor (residual soft tissue mass measuring ) will receive 21.6 Gy EBRT to the postinduction chemotherapy, pre-operative primary tumor volume and an additional boost of 14.4 Gy EBRT to the gross residual tumor (total dose 36 Gy to gross residual tumor volume).
Timing of Radiation Therapy: Radiation will be given after stem cell transplantation and should start no sooner than 28 days post transplant. Organ toxicity within radiation field should have resolved or meet the protocol criteria for starting radiation therapy that are included in Section 17.0. It is recommended to start radiation therapy within 42 days after stem cell transplant.
ANBL0032 demonstrated an improvement in EFS for patients who recieved ch14.18 immunotherapy plus cytokines in addition to cis-RA. Patients should be encouraged to participate in clinical trials of ch14.18 immunotherapy (ie. ANBL0032 or ANBL0931). Patients ineligible for immunotherapy or who decline participation will remain on ANBL0532 for Maintenance Phase using cis-RA alone.
COG #
Maintenance Phase: Post-transplant Maintenance therapy with cis-RA daily for 14 days every 28 days repeated for 6 months will be administered. To begin after completion of radiation therapy and criteria below are met.
Patient name or initials
DOB
Criteria to Begin Each Cycle of Isotretinoin (Accutane) (13-cis-Retinoic Acid)
Prior to each cycle a patient must have: ALT X normal, Skin toxicity no greater than Grade 1; Serum Triglycerides ; No hematuria and/or proteinuria on urinalysis; Serum creatinine < 1.5 times normal value based upon age and gender (see Section 3.2.4.1). This Therapy Delivery Map is one page.
DRUG
ROUTE
DOSAGE Patients
DOSAGE Patients > 12kg
DAYS
IMPORTANT NOTES
OBSERVATIONS
Isotretinoin (ISOT) (Accutane)
Orally
day divided BID
day divided BID
Days 1-14
Round up to nearest 10 mg
a. Physical, Ht, Wt
b. CBC with diff and platelets
c. Electrolytes, BUN, Cr, Ca, Phos, Mg
d. ALT, AST, Bilirubin, Albumin, Triglyceride, Urinalysis
e. Cis-RA PK (Section 15.7)
f. Bilateral BMA/Bx
g. BM PCR (Section 15.4)
h. Peripheral Blood for Immune recovery (Section 7.1.3)
i. Tumor Imaging, Bone scan, MIBG (Section 7.1.3)
j. MRI Spine & Neuro Eval (tumors with intraspinal extension only)
k. VMA, HVA (if elevated at diagnosis) 1. ECG, MUGA/ECHO, GFR, Audiogram/BAER (see Section 7.1.3) m. Peripheral Blood PCR (Section 15.6) OBTAIN OTHER STUDIES AS REQUIRED FOR GOOD PATIENT CARE
Therapy Delivery Map
Cycle
of 6
Ht
cm
Wt
kg
BSA
Date Due
Date Given
Day
ISOT mg
Studies
Comments (Include any held doses, or dose modifications)
Enter calculated dose above and actual dose administered below
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
ANBL0532
5.0 DOSE MODIFICATIONS FOR TOXICITIES
All toxicities should be graded according to the Common Terminology Criteria for Adverse Events (CTCAE) v3.0 (Starting July 2011, it will be graded according to CTCAE V.4.0).
5.1 Myelosuppression During Induction
5.1.1
Dose adjustments will be made based on the neutrophil count and platelet count on Day 29 of each cycle. Following any cycle, if the and/or platelet count is on Day 22, delay next cycle until recovery occurs or meets criteria to continue based on bone marrow tumor involvement (see below). If patient recovers to and platelets by Day 29, proceed with next cycle at full dose. If patient has not met hematopoietic recovery criteria on or before Day 29, perform bone marrow aspirate and biopsy. Proceed based on hematopoietic recovery criteria (see Section 5.1.2).
5.1.2 Hematopoietic Recovery Criteria
If the marrow is positive for tumor at diagnosis or at last evaluation, still contains tumor and is normocellular or mildly hypocellular with trilineage hematopoiesis proceed with the next cycle of chemotherapy without alteration in dose regardless of ANC and platelets. If the marrow has no tumor and is severely hypocellular without tri-lineage hematopoiesis, delay the next cycle of therapy until the ANC and platelets .
If recovery occurs between Day 30-43 for any cycle of Induction, reduce the doses of all drugs except vincristine by .
If recovery occurs after Day 43 of any cycle, reduce drug doses by , except for vincristine.
5.2 Hematuria During Induction
For Cycles 1 and 2: If microscopic ( abnormal urinalyses during a cycle of therapy with < Grade 2 hematuria) or gross hematuria occurs after Induction Cycle 1 cyclophosphamide, give MESNA with Induction Cycle 2 cyclophosphamide as follows: MESNA (or if ) with cyclophosphamide infusion, then MESNA (or if ) IV over 15 minutes at Hours 4 and 8 from start of cyclophosphamide infusion. If hematuria resolves prior to start of cycle 4 cyclophosphamide, administer cyclophosphamide and mesna in cycles 4 and 6 without modification.
For Cycles 4 and 6: If microscopic ( abnormal urinalyses during a cycle of therapy with < Grade 2 hematuria) or gross hematuria occurs after Induction Cycle 4 cyclophosphamide, give mesna as a 24 hour continuous infusion with Induction Cycle 6 cyclophosphamide as follows: MESNA (or 18.7 if ) with each cyclophosphamide infusion, then MESNA (or if kg ) in required fluid over 18 hours after cyclophosphamide infusion completed.
If Grade 3 or 4 hematuria occurs following a cycle of cyclophosphamide, do not give another cycle of cyclophosphamide, topotecan (CT) or cyclophosphamide, doxorubicin and vincristine (CDV) until hematuria resolves to Grade 2 or less. If patient is due to begin next cycle of cyclophosphamide containing chemotherapy prior to resolution of hematuria to Grade 2, substitute cisplatin and etoposide cycle. Make notation of substitution on data forms. The intent of Induction is to give a total of 2 cycles each of CT, cisplatin/etoposide and CDV, therefore if substitution of cisplatin/ etoposide is made for CDV cycle or a CT cycle, make-up this missed cyclophosphamide-containing cycle later in therapy. If gross hematuria from cyclophosphamide recurs, delete cyclophosphamide from subsequent cycles.
ANBL0532
5.3 Renal Toxicity During Induction
5.3.1 Cisplatin
No dose reductions in cisplatin will be made for a decrease in the baseline GFR or creatinine clearance as long as the value remains . If the serum creatinine increases during a cycle of cisplatin-containing chemotherapy, or increased to greater than maximum serum creatinine for age as listed in table below, omit the remainder of the cisplatin from that cycle.
Age
Maximum Serum Creatinine (mg/dL)
Male
Female
1 month to < 6 months
0.4
0.4
6 months to < 1 year
0.5
0.5
1 to < 2 years
0.6
0.6
2 to < 6 years
0.8
0.8
6 to < 10 years
1
1
10 to < 13 years
1.2
1.2
13 to < 16 years
1.5
1.4
years
1.7
1.4
The threshold creatinine values in this Table were derived from the Schwartz formula for estimating GFR (Schwartz et al. J. Peds, 106:522, 1985) utilizing child length and stature data published by the CDC.
If GFR or creatinine clearance is prior to cisplatin/etoposide cycle substitute CDV cycle. Make notation of substitution on data forms. The intent of Induction is to give a total of 2 cycles of CT, 2 cycles of cisplatin/etoposide, and 2 cycles of CDV, therefore if substitution of CDV cycle is made for cisplatin/etoposide cycle, give the cisplatin/etoposide cycle later in therapy. Omit further cycles of cisplatin therapy if GFR or creatinine clearance remains .
5.3.2 Cyclophosphamide, Doxorubicin, Vincristine, Topotecan and Etoposide
No dose reductions in cyclophosphamide, doxorubicin, vincristine, topotecan or etoposide are necessary for decrease in creatinine clearance.
5.4 Cardiac Toxicity During Induction
5.4.1 For Change In Ejection/Shortening Fraction
If the cardiac ejection fraction falls below or shortening fraction below and patient is asymptomatic following a cycle of doxorubicin, repeat the study in one week. If the ejection fraction or shortening fraction remains abnormal one week later proceed as follows:
If cardiac toxicity occurs prior to Cycle 4, substitute cycle of cisplatin/etoposide for CDV. Make notation of substitution on data forms. The intent of Induction is to give a total of 2 cycles of CDV and 2 cycles of cisplatin/etoposide, therefore if cisplatin/etoposide cycle substituted for CDV, give CDV cycle later in therapy if possible. If cardiac function does not return to normal prior to cycle 6 or is initially noted to be abnormal prior to cycle 6 scheduled CDV, omit doxorubicin.
5.4.2 For Symptomatic Congestive Heart Failure (CHF)
If at any time, the patient develops Grade 3 congestive heart failure or dysrhythmia or any Grade 4 cardiac toxicity not related to underlying infection or metabolic abnormality, omit doxorubicin from all subsequent cycles. If cardiac toxicity is resolved to Grade 2 congestive heart failure or dysrhythmia,
decrease the dose of cyclophosphamide to for the next cycle containing cyclophosphamide. If this dose of cyclophosphamide is tolerated without Grade 2 congestive heart failure or dysrhythmia, then administer full dose of cyclophosphamide in subsequent cycles of chemotherapy.
Congestive Heart Failure will be referred as "Heart Failure" per CTCAE v4.0 followed starting July , 2011.
5.4.3 For Dysrhythmia
If the patient develops Grade 2 cardiac dysrhythmia as defined in the Common Toxicity Criteria, repeat in one week. If Grade 2 toxicity resolves to Grade 0 or 1 toxicity, patient may continue on therapy without chemotherapy dose alterations.
If Grade 2 toxicity occurs prior to Cycle 4, substitute cisplatin/etoposide for CDV cycle until dysrhythmia resolves. Make a notation of chemotherapy substitution on data form. The intent of Induction is to give a total of 2 cycles of CDV and 2 cycles of cisplatin/etoposide, therefore if cisplatin/etoposide cycle substituted for CDV, give CDV cycle later in therapy. If dysrhythmia symptoms occur prior to Cycle 6, proceed with cyclophosphamide, vincristine but omit doxorubicin.
5.4.4 Hypertension
Hypertension due to neuroblastoma will not be considered reason for removal from protocol therapy or alteration in chemotherapy doses.
5.5 Hepatotoxicity During Induction
If direct bilirubin is prior to Cycle 4 chemotherapy, substitute cisplatin/etoposide for CDV cycle. If direct bilirubin is prior to Cycle 6 chemotherapy, omit doxorubicin and vincristine. If direct bilirubin is but (Grade 3 toxicity) prior to Cycle 4 or 6 chemotherapy, reduce doxorubicin and vincristine dose by .
5.6 Gastrointestinal Toxicity During Induction
5.6.1 Mucositis
If patient develops Grade 3 or 4 mucositis that resolves to < Grade 2 by Day 22-29 of next cycle, no dose adjustments will be made in chemotherapy. If patient develops Grade 3 or 4 mucositis that is NOT attributable to infectious etiology AND recovery to < Grade 2 occurs between Day 30-43 for any cycle of Induction, reduce the dose of doxorubicin or etoposide in the next 2 cycles of chemotherapy by . If subsequent chemotherapy tolerated without recurrence of Grade 3 or 4 GI toxicity then resume full doses of chemotherapy agents in all subsequent cycles of induction.
If patient develops Grade 3 or 4 mucositis that is NOT attributable to infectious etiology AND recovery to < Grade 2 occurs after Day 43 of any cycle, reduce dose of doxorubicin or etoposide in the next 2 cycles of chemotherapy by . If subsequent chemotherapy tolerated without recurrence of Grade 3 or 4 GI toxicity then escalate dose by in subsequent cycles of induction.
If patient develops mucositis that requires intubation for airway management or if patient develops grade 4 typhlitis or other grade 4 gastrointestinal toxicity hold subsequent chemotherapy until toxicity resolved to < Grade 2. If the toxicity resolves to < Grade 2 by Day 43, proceed with next 2 cycles of chemotherapy but reduce dose of doxorubicin or etoposide by . If recovery to Grade 2 occurs after Day 43 of any cycle, reduce dose of doxorubicin or etoposide in the next 2 cycles of chemotherapy by . If subsequent chemotherapy tolerated without recurrence of Grade 3 or 4 GI toxicity then escalate dose by in subsequent cycles of induction.
ANBL0532
5.6.2 Diarrhea
If patient develops severe diarrhea (Grade 3 or 4) attributable to chemotherapy and not underlying infection (i.e. C. difficile), that resolves by Day 22-29 of cycle, no dose adjustments will be made in chemotherapy. If recovery to < Grade 2 occurs between Day 30-43 for any cycle of Induction, reduce the dose of doxorubicin or etoposide in next cycle of chemotherapy by . If subsequent chemotherapy tolerated without recurrence of Grade 3 or 4 GI toxicity then resume full doses of chemotherapy agents in all subsequent cycles of induction. If recovery to < Grade 2 occurs after Day 43 of any cycle, reduce dose of doxorubicin or etoposide in the next cycle of chemotherapy by . If subsequent chemotherapy tolerated without recurrence of Grade 3 or 4 GI toxicity then escalate dose by in subsequent cycles of induction.
5.7 Ototoxicity During Induction
For an inner ear/hearing toxicity Grade 3 , decrease cisplatin dose by for subsequent cycles. If loss extends below 2000 Hz , delete further cisplatin/etoposide cycles. If cisplatin is deleted, then complete total of 2 cycles of CDV, then proceed to consolidation therapy. Make notation of cisplatin deletion on data form.
5.8 Neurologic Toxicity During Induction
If severe peripheral neuropathy (vocal cord paralysis, inability to walk or perform usual motor functions) or ileus develops from vincristine, vincristine therapy should be stopped or withheld until the ileus resolves or the peripheral neuropathy improves. Restart vincristine at dose and escalate by if tolerated with next course. If neuropathy recurs on escalating dose, return to previously tolerated dose once neuropathy improved.
5.9 Allergic Reactions
5.9.1 Etoposide
Etoposide allergic reactions may be managed with pre-medications such as diphenhydramine IV (maximum single dose 50 mg ), ranitidine IV (maximum single dose 50 mg ) and Hydrocortisone IV and by slowing the rate of the infusion. For those reactions which are unable to be controlled with pre-medication and the slowing of the rate of etoposide infusion, etoposide phosphate may be substituted in the same dose and at the same rate. Pre-medication for etoposide phosphate is recommended.
5.9.2 Cisplatin and Carboplatin
Platinum allergic reactions may be managed with pre-medications such as diphenhydramine IV (maximum dose 50 mg ), ranitidine IV (maximum single dose 50 mg ) and Hydrocortisone 1-4 mg/kg IV.
5.10 Other toxicities During Induction
For any Grade 3 or 4 toxicity not mentioned above, the treatment should be withheld until patients recover to Grade 2 or less toxicity. For any non-hematologic Grade 3 or 4 organ toxicity attributed to chemotherapy AND not related to underlying infection or metabolic derangement that is not discussed in Sections 5.1-5.8, and resolves to < Grade 2 by Day 43, reduce the subsequent dose of that chemotherapy agent by . For any non-hematologic Grade 3 or 4 toxicity attributed to chemotherapy AND not related to underlying infection or metabolic derangement that is not discussed in Sections 5.1-5.8 resolves to < Grade 2 greater than Day 43, reduce the subsequent dose of that chemotherapy by .
5.11 Dose Modifications for 13-cis-Retinoic Acid (cis-RA) Therapy
5.11.1
A dose reduction of (to day day if child weighs ) for subsequent cycles should be made for the occurrence of any Grade 3 or 4 toxicities EXCLUDING: Grade 3 or 4 hematologic, Grade 3 hepatic, Grade 3 nausea, Grade 3 vomiting, or Grade 3 fever. If the same Grade 3 or 4 toxicity recurs at a dose reduction, then decrease dose another (to day 3.33 day if child weighs . If the same Grade 3 or 4 toxicity recurs after two dose reductions, and toxicity cannot be attributed to an alternative etiology, then withhold further therapy.
5.11.2
It has been reported (rarely) that some patients treated with cis-RA develop new areas of abnormal uptake on bone scan, likely due to increased bone resorption. If such changes occur during retinoic acid phase in absence of other evidence of tumor recurrence do not report as progressive disease.
5.11.3
If criteria (Section 4.8.1) to begin next cycle are not met by the date cycle is due to begin, delay cycle for one week. If criteria still not met, hold therapy until criteria are met, and treat at dose reduction ( 120 day or day if child weighs ). An additional dose reduction to day ( 3.33 day if child weighs ) should occur if criteria are not met within one week after due date for subsequent cycles.
5.11.4
If serum creatinine increases by in any cycle of therapy, creatinine clearance or GFR should be done prior to starting next cycle, and a urinalysis. If creatinine clearance and/or GFR are reduce cis-RA by . If serum creatinine continues to increase or GFR decreased to then withhold further therapy.
5.11.5
If patient > Grade 1 develops hematuria, proteinuria, and/or hypertension during any cycle of therapy, hold medication until resolves to Grade 1 then reduce cis-RA dose by .
5.11.6
For localized cheilitis, apply topical vitamin E to lips for subsequent cycles. If this does not control symptoms sufficiently to allow sufficient oral intake, then decrease dose by day or 4 day if child weighs ).
5.11.7
If serum triglycerides are when next cycle is due, delay starting therapy for two weeks. If still , then start patient on medical therapy for serum triglyceride reduction (consider cardiology consultation) and begin cycle at previous cis-RA dosage. If serum triglycerides are < 300 by time subsequent cycle is due, then continue at same dosage cis-RA. If triglycerides are still > after one cycle on medical therapy, then reduce cis-RA acid dosage by for subsequent cycles.
6.0 DRUG INFORMATION
See the consent document for toxicities. All other information is available on the COG website in the manual titled "Drug Information for Commercial Agents used by the Children's Oncology Group" at: https://members.childrensoncologygroup.org/prot/reference_materials.asp under Standard Sections for Protocols.
Children's Oncology Group
7.0 EVALUATIONS/MATERIAL AND DATA TO BE ACCESSIONED
All baseline studies must be performed prior to starting protocol therapy unless otherwise note below.
Timing of protocol therapy administration, response assessment studies, and surgical interventions are based on schedules derived from the experimental design or on established standards of care. Minor unavoidable departures (up to 72 hours) from protocol directed therapy and/or disease evaluations (and up to 1 week for surgery) for valid clinical, patient and family logistical, or facility, procedure and/or anesthesia scheduling issues are acceptable per COG administrative Policy 5.14 (except where explicitly prohibited within the protocol).
7.1 Required Clinical, Laboratory and Disease Evaluations
7.1.1 Required Observations Pre-Treatment and During Induction
Observation
Pre-Treatment
Prior to Each Cycle
During Cycle 1, 2
Following Cycle 2
At PBSC Harvest
Prior to Surgery
End of Induction
At Relapse
Physical Exam
X
X
X
Height, Weight
X
X
X
CBC with diff/platelets
X
X
Electrolytes, BUN, Cr., Ca, Phos, Mg
X
X
ALT, AST, Bilirubin, Urinalysis, albumin
X
X
X
ECG and MUGA or ECHO
X
X
GFR or Creatinine Clearance
X
Audiogram or BAERs
X
X
Bilateral BM Asp/Bx
X
X
BM PCR
BM ICC
Peripheral Blood PCR
Peripheral blood for immune assessments
X
Topotecan pharmacokinetics
X
Pharmacogenetics
X
PBSC PCR
Tumor Imaging
X
X
X
X
X
MRI Spine and Neurologic Evaluation
X
X
X
Bone Scan
X
X
MIBG
X
Catecholamines (VMA, HVA)
X
Pregnancy Test
X
. Camera method IS NOT allowed as measure of GFR prior to or during Consolidation therapy if GFR/ creatinine clearance is .
BM studies to be done prior to starting Cycle 3 (This sample may be obtained prior to PBSC harvest for convenience if patient is undergoing anesthesia for apheresis line placement. DO NOT NEED results to proceed to PBSC collection). Send bone marrow and if biopsy performed, send sample of fresh and snap frozen tumor. Ship per ANBL00B1 guidelines. Label "relapse" specimen.
Diagnosis & End of Induction: Obtain 10 cc of bone marrow ( 5 cc from each side) in sodium heparin tube. Following cycle 2 obtain 0.5 cc from a single BM site for PCR only. See Section 15.4 for details regarding bone marrow immunocytochemistry and PCR analyses.
Obtain 2 mL peripheral blood in sodium heparin or EDTA tube. See section 15.6 for details.
Obtain 20 cc peripheral blood in sodium heparin tubes prior to initiation of chemotherapy. See Section 15.8 for details Collect 2 cc in green top tube 15 minutes after completion of topotecan infusion on day 1 of Cycles 1 and 2. See Section 15.3 for details and ordering of sample collection kit.
Collect one time sample of 10 cc in purple top tube (ETDA tube) prior to start of therapy (preferable) or when patient is not leukopenic ( ). See section 15.2 for details.
Collect 1 cc of PBSC into sodium heparin tube and then transfer to PAX gene tubes. See Section 15.5 for details. Tumor imaging CT/MRI as needed for optimum visualization of all areas of bulk tumor (primary and tastases). CT/MRI required prior to surgical resection. Repeat imaging of surgical sites within 4-6 weeks post-operatively (can be the same as end of induction scans if this is within 4-6 weeks of surgical resection). If patient has radiographic CR at end of induction submit these scans for Central Review.
Obtain for tumors with intraspinal extension only. See Section 13.4 and Appendix III for details. Obtain if positive at diagnosis and results discordant with MIBG scan or tumor MIBG non-avid MIBG scan can be performed within 2 weeks of starting chemotherapy if it is not possible to obtain scan prior to starting chemotherapy. Repeat MIBG later in therapy only if positive at diagnosis. I MIBG scan preferred, see section 16.4 for details.
Repeat catecholamine levels if elevated at diagnosis.
Perform for all females of childbearing age.
PLEASE NOTE THAT END OF INDUCTION (Section 7.1.1) AND PRIOR TO CONSOLIDATION (Section 7.1.2) TESTS REQUIRED ARE THE SAME AND ARE NOT TO BE PERFORMED TWICE UNLESS SPECIFIED
7.1.2 Required Observations During Consolidation Therapy, Regimen A and Regimen B
Observation
Prior to Consolidation
During HSCT #1 chemo Regimen A and B
During Consolidation HSCT #1 Regimen A and B
Prior to HSCT # 2 Regimen B
During Consolidation HSCT #2 Regimen B
Post HSCT #1 (regimen A) or HSCT #2 (regimen B)
End of Consolidation Phase (after radiation)
At Relapse
Physical Exam, Ht., Wt.
X
X
X
X
CBC with diff./platelets
X
X
X
Electrolytes, BUN, Cr
X
X
X
Triglycerides
X
, Phos
X
X
X
AST, ALT, Bilirubin, Albumin
X
X
X
Urinalysis
X
X
X
ECG and MUGA or ECHO
X
X
GFR or Creatinine Clearance
X
X
Audiogram or BAERs
X
Bilateral BM Asp/Bx
X
BM PCR
X
BM ICC
Peripheral Blood PCR
X
Peripheral blood for immune assessments
Peripheral blood for immune recovery
Tumor Imaging
X
X
MRI Spine and Neurologic Assessment
X
Bone Scan
X
X
MIBG
X
X
Catecholamines VMA, HVA
X
1 Must be performed within the 2 weeks prior to start of consolidation therapy unless otherwise noted.
2 Daily during consolidation chemotherapy administration and day of PBSC infusion. CBC daily until neutrophil engraftment, then weekly through radiation therapy.*
3 Obtain every other day if stable. Obtain on day of PBSC infusion*.
4 Obtain twice weekly if stable*
5 No restriction on method for GFR calculation. If a creatinine clearance is performed prior to beginning consolidation therapy after completing induction and /or before Regimen B HSCT #2 if applicable and the result is then the test must be repeated and a GFR performed. The GFR must be obtained by blood sampling method or iothalamate clearance method for all patients with creatinine clearance . Camera method is NOT allowed as measure of GFR prior to or during Consolidation therapy for patients whose GFR/creatinine clearance is .
6 Must be performed within 1 month prior to start of consolidation therapy. These observations should have been performed at the end of induction and do not need to be repeated unless consolidation begins weeks after the end of induction.MIBG obtained only if positive at diagnosis. Repeat catecholamine levels if elevated at diagnosis.
7 Send bone marrow and if biopsy performed, send sample of fresh and snap frozen tumor. Ship per ANBL00B1 guidelines. Label "relapse" specimen.
8 Obtain 10 cc of bone marrow ( 5 cc from each side) in sodium heparin tube. See section 15.4 for details regarding bone marrow immunocytochemistry (ICC) and PCR analyses.
9 Obtain 2 ml peripheral blood in sodium heparin or EDTA tube. See section 15.6 for details.
10 Obtain 20 cc peripheral blood in sodium heparin tubes months after final HSCT procedure, preferably prior to initiation of Accutane therapy or at the time of enrollment on ANBL0032 or ANBL0931. See section 15.8 for details.
11 Send CBC with differential and measurement by flow cytometry of peripheral blood CD3, CD4 and CD8 at 1,3 and 6 months post the final HSCT procedure (HSCT on Regimen A or HSCT #2 on Regimen B). To be performed at local institutions.
12 Tumor imaging CT/MRI as needed for optimum visualization of all areas of prior or persistent bulk tumor (primary and metastases).
13 Obtain for tumors with intraspinal extension only. See section 13.4 and Appendix III for details.
14 Bone scan required only if positive at diagnosis and results discordant with MIBG scan or MIBG non-avid tumor
15 MIBG scans performed only if the tumor is MIBG avid at diagnosis. I-123 MIBG scans is preferable, see section 16.4.
16 Obtain catecholamine levels if elevated at diagnosis
* Please follow your institutional guidelines. In the absence of such guidelines you may follow the recommendations listed above in footnotes 2, 3 and 4.
PLEASE NOTE THAT END OF INDUCTION (Section 7.1.1) AND PRIOR TO CONSOLIDATION (Section 7.1.2) TESTS REQUIRED ARE THE SAME AND ARE NOT TO BE PERFORMED TWICE UNLESS SPECIFIED
7.1.3 Required Observations During 13-cis-Retinoic Acid (Maintenance)
Observation
Prior to each cycle of cis-RA
Before Cycle cis-RA
End of Therapy
At Relapse
Physical Exam, Ht., Wt.
X
X
CBC with diff./platelets
X
X
Electrolytes, BUN, Cr
X
X
Triglycerides
X
X
, Phos
X
X
AST, ALT, Bilirubin, Albumin
X
X
Urinalysis
X
X
ECG and MUGA or ECHO
X
GFR or Creatinine Clearance
X
Audiogram or BAERs
X
Bilateral BM Asp/Bx
X
X
X
BM PCR
X
Peripheral Blood PCR
X
Peripheral blood for immune recovery
X
Cis-RA PK
Tumor Imaging
X
X
X
MRI Spine and Neurologic Assessment
X
Bone Scan
X
X
X
MIBG
X
X
X
Catecholamines VMA, HVA
1 Send bone marrow and if biopsy performed, send sample of fresh and snap frozen tumor. Ship per ANBL00B1 guidelines. Label "relapse" specimen.
2 Obtain 10 cc of bone marrow ( 5 cc from each side) in sodium heparin tube. See section 15.4 for details regarding immunohistochemistry (ICC) and PCR analyses.
3 Obtain 2 ml peripheral blood in sodium heparin tube. See section 15.6 for details.
4 Send CBC with differential and measurement of and CD 8 by flow cytometry at 1,3 and 6 months post the final HSCT procedure (HSCT on Regimen A or HSCT #2 on Regimen B)
5 Collect 5 cc blood into sodium heparin tube wrapped in foil 4 hours after cisRA dose on Day 14 of Cycle 1. See section 15.7 for details.
6 Tumor imaging CT/MRI as needed for optimum visualization of all areas of prior or persistent bulk tumor (primary and metastases).
7 Obtain for tumors with intraspinal extension only. See Section 13.4 and Appendix III for details.
8 Bone scan required only if positive at diagnosis and results discordant with MIBG scan or MIBG non-avid tumor
9 MIBG scans performed only if the tumor is MIBG avid at diagnosis. I-123 MIBG scans is preferable, see Section 16.4.
10 Obtain catecholamine levels if elevated at diagnosis
7.2 Required Observations During Follow-up After Completion of 13-cis-Retinoic Acid Therapy (for All Patients)
Count time 0 as date of disease evaluation after completion of last cycle of cis-RA.
Observation
3 Months
6 Months
9 Months
1 Year
1.5 Year
2 Years
2.5 Years
3 Years
3.5 Years
4 Years
4.5 Years
5 Years
Annually After 5 Years
At Relapse
Physical Exam
X
X
X
X
X
X
X
X
X
X
X
X
X
Height, Weight
X
X
X
X
X
X
X
X
X
CBC with differential and platelets
X
X
X
X
X
X
X
X
X
X
X
X
X
MUGA or ECHO, ECG
X
X
Bilateral
Tumor Imaging
X
X
X
X
X
X
X
X
X
Bone Scan
X
X
X
X
X
X
X
X
MIBG
X
X
X
X
X
X
X
X
Catecholamines
X
X
X
X
X
X
X
X
X
X
X
X
TSH, T4, Pulmonary Function tests
Perform physical exam and CBC monthly for one year after transplant (recommended).
Perform if positive at diagnosis.
Bone marrow evaluations and MIBG scans only if positive after completion of Cis-retinoic acid therapy.
Perform as scheduled, then as clinically indicated. Bone scan required only if positive at diagnosis and results discordant with MIBG scan or MIBG non-avid tumor. For patients with tumors with intraspinal extension obtain MRI of spine and perform neurologic assessments per Appendix III at months and annually through year 5 .
If abnormal, repeat at 2 years and as needed. If child is years when tested as directed, an additional test should be performed when child becomes age 5 .
Send bone marrow and if biopsy performed, send sample of fresh and snap frozen tumor. Ship per ANBL00B1 guidelines. Label "relapse" specimen.
Perform PFTs only if child has pulmonary symptoms and if child is years. Perform thyroid function tests only for patients with poor linear growth or other concern about poor thyroid function. (recommended)
Patient's clinical status will be tracked annually through 10 years after enrollment onto study.
8.0 OFF PROTOCOL THERAPY CRITERIA AND OFF STUDY CRITERIA
8.1 Off Protocol Therapy Criteria
8.1.1 During Induction Therapy
a) Patient with progressive disease at end of Induction therapy will be off protocol therapy. Patients with mixed response, stable disease or persistent tumor in bone marrow by morphologic evaluation may remain on protocol therapy as per investigator preference. These patients are encouraged to come off protocol therapy for entry onto available COG Phase I and Phase II studies.
b) Patient develops any Grade 4 organ toxicity not related to underlying infection or metabolic derangement that fails to resolve to < Grade 2 by 8 weeks from last cycle of chemotherapy
EXCEPT:
Patients with Grade 4 toxicities of fever, infection, ileus, nausea, transaminases, renal electrolyte wasting, vomiting, diarrhea, and stomatitis (not requiring intubation) who may continue on therapy if these toxicities resolve or are controlled with medication by Day 56 of cycle.
c) Unable to obtain adequate stem cell product for transplant.
d) Patient with at the end of induction. It is to be noted that GFR must be confirmed using nuclear blood sampling method or iothalamate clearance method ONLY and NOT camera method.
e) Patients with left ventricular cardiac dysfunction at the end of induction with ejection fraction or shortening fraction .
8.1.2 During Stem Cell Transplant
a) Patients who develop progressive disease (PD) at any time after stem cell transplant are off protocol therapy and may receive alternate therapy. If the patient has PR or SD after stem cell transplant i.e., questionable residual tumor, a biopsy should be done to obtain histological diagnosis, if possible, to determine if there is residual disease. If active malignant tumor remains, notify the Study Chair prior to additional therapy. Surgical removal is not encouraged unless it would alter therapy. Additional local irradiation should also be considered.
8.1.3 Treatment Completed Per Protocol
Patients who are off protocol therapy are to be followed until they meet the criteria for Off Study (see below). Follow-up data will be required unless consent was withdrawn.
8.1.4 Entire therapy
a. Patient/Parent refuses further treatment (including patient/parent who refuses randomization)
b. Patient/parent withdraws consent for any further data submission.
c. Physician determines it is in patient's best interest.
d. Development of a 2nd malignancy
8.2 Off Study Criteria
a) Death
b) Lost to follow-up
c) Entry onto another COG study with tumor therapeutic intent (e.g., alternative maintenance therapy protocol or at recurrence)
d) Withdrawal of consent for further data submission
e) Tenth anniversary of study entry
9.0 STATISTICAL CONSIDERATIONS
Overview
This study will address the issue of whether an additional myeloablative consolidation results in better EFS than a single myeloablative consolidation, sufficient to justify the additional toxicity and duration of therapy.
9.1 Statistical Design
Randomization
This is a prospective study with a delayed two-arm randomization. With delayed randomization, early induction failures will be excluded from the EFS comparison and will prevent dilution of treatment effect. Randomization will be stratified by stage and MYCN status (the two factors most highly prognostic of outcome in neuroblastoma), and also by the end of induction response (CR/VGPR vs PR vs MR/NR).
Timing: Randomization will occur after the completion of 6 cycles of induction therapy (including surgery) prior to transplant.
Eligibility to proceed to Consolidation Therapy/Transplant: Only patients who have a minimum PBSC harvest of cells (minimum of cells transplant), who have adequate organ function, who do not have progressive disease at end of induction AND who have consented to Consent Part 2 are eligible for transplantation.
Eligibility for randomization: Patients must first be eligible to proceed to transplant in order to be eligible for randomization. Patients will be randomly assigned to either single myeloablative therapy (Regimen A) with a carboplatin, etoposide, melphalan (CEM) preparative regimen and autologous PBSC rescue (representing the standard arm of the study), to the experimental arm of tandem myeloablative therapy (Regimen B) with the regimen used in the COG ANBL00P1 (thiotepa and cyclophosphamide followed by CEM).
Nonrandom Assignment to Single Transplant
The following patients will be nonrandomly assigned to single myeloablative transplant (Regimen A):
Patients who are 365 to 547 days of age ( 12 - 18 months) with Stage 4, MYCN nonamplified tumor with any of the following potentially unfavorable biologic features:
unfavorable histopathology
diploid DNA content
indeterminant histopathology
indeterminate ploidy
Patients who are greater than 547 days of age with Stage 3, MYCN nonamplified tumor with either of the following:
unfavorable histopathology
indeterminant histopathology
Treatment Groups
10 = Single HSCT (CEM)
20 = Tandem HSCT (TC, then CEM) Induction
HSCT - Hematopoietic Stem Cell Therapy
Strata
Strata (cont.)
01 = non-randomly assigned to Trt 01:AT-CEM
09 = stage 3; MYCN amp; PR
02 = stage 4s; amp; CR/VGPR
10 = stage 4; not amp or indeterminant; PR*
03 = stage 2a/2b; amp; CR/VGPR
11 = stage 4; amp; PR
04 = stage 3; MYCN amp; CR/VGPR
stage 4s; amp; MR/NR
05 = stage 4; not amp or indeterminant;
13 = stage 2a/2b; MYCN amp; MR/NR
CR/VGPR*
06 = stage 4; amp; CR/VGPR
stage 3; amp; MR/NR
stage 4s; amp; PR
15 = stage 4; not amp or indeterminant; MR/NR*
stage 2a/2b; amp; PR
stage 4; amp; MR/NR
* - Excludes patients 12-18 months old
CR - Complete response
VGPR - Very Good Partial Response
PR - Partial Response
MR - Mixed Response
NR - No Response
9.2 Patient Accrual and Expected Duration of Trial
9.2.1 Accrual
Based on A3973 and CCG-3891, annual accrual was expected to be about 140 patients/yr. Of these, are expected to go off protocol therapy or off study prior to randomization due to physician discretion, tumor progression, death, or inability to achieve the cells needed to proceed to randomization/transplantation. Of the remaining 98 patients/year, 13 are estimated to be stratum 01 patients who are non-randomly assigned to receive a single transplant (AT). Furthermore, the delayed timing of randomization and the criteria to discontinue protocol therapy for those with progressive disease should result in a very low rate for refusal of randomization (about ). Therefore, the original estimate was for 81 patients/year (69/year of which are Stage 4) to be randomized. However, as of the second quarter of 2011, the annual accrual rate has reached 230 patients/year and the randomization rate is , so the new estimate of the randomized patient rate is 115 patients/year.
In the original plans, a total of 329 randomized patients were needed for sufficient power for an analysis of the primary EFS treatment comparison. At an accrual rate of 81 patients per year, 329 would be accrued in about 4.1 years, plus 3 of follow-up, for a total study duration of about 7 years. Including stratum 01 patients and patients who go off protocol therapy or off study prior to randomization, the total study accrual was originally planned as 574 patients ( 140 /year for 4.1 years). Based on the observation of higher EFS rates than anticipated in A3973 and ANBL0032, the power calculations below have been revised, and a new total enrollment of up to 664 patients will be required.
To increase the homogeneity of the patient cohort, patients will be strongly encouraged to enroll on ANBL0032 or ANBL0931 after ANBL0532. The DSMC will be kept apprised of the proportion of patients who enroll on ANBL0032 or ANBL0931, overall and by ANBL0032 treatment arm. (Prior to closure of randomization, ANBL0032 randomization was stratified by ANBL0532 treatment arm.)
9.2.2 Power Calculations
In the original plan: Power for EFS treatment comparison: randomized patients can detect a difference in the 3-year EFS rate starting from the time of randomization (from for AT versus for AT1+AT2, which is a hazard ratio of 0.7 ) with power in a one-sided logrank test at a 0.05 significance level. The -year EFS was chosen for the control group in this study based on the national trend for improvement in local control rates, use of more dose intensive therapy, and better supportive care occurring since the design of 3891 with -year EFS. A 3 year follow-up provides time for sufficient events for power and allows for some normalization of the EFS time posttransplant for 1 versus 2 transplants. The more patient-years of follow-up there are, the smaller the proportion of time represented by the additional EFS time post-transplant of the tandem transplant patients.
For the new plan (Aug 2011):
The new control arm 3 -year EFS will be , starting from the time of randomization. In order to detect a difference in the 3-year EFS rate starting from the time of randomization (from for AT1 versus for AT1+AT2) with power in a one-sided logrank test at a significance level, and taking into account a crossover rate from AT1+AT2 to AT1, a total of 332 randomized patients will be required. (This results in a change from the originally planned hazard ratio (HR) [AT1+AT2: AT1] of 0.7 to a new HR of 0.64.) In order to accrue 332 randomized patients, we will need to enroll 664 patients (which assumes that the randomization rate might drop to as low as ).
The choice was made to limit accrual duration to 4 years, and so the sample size of stage 4 patients provides too little power for the study to be designed/powered on the basis of this subgroup. A year EFS difference can be detected with power based on the 276 stage 4 patients expected to be randomized.One-hundred and seventy-seven events are expected to be observed within the cohort of randomized patients after completion of 3 years follow-up after the last patient is accrued.
9.3 Statistical Analysis Methods
9.3.1 Endpoints
9.3.1.1 Primary Endpoint
The primary end point to determine benefit of a myeloablative consolidation procedure is the eventfree survival (EFS) rate. A potential inherent bias exists due to the longer duration of therapy for patients who get two myeloablative consolidations. Ideally the duration of treatment for two randomized groups would be the same, such that all patients complete their treatment at about the same time from randomization. However, in this study the patients randomized to two transplants will take longer to finish their treatment than those randomized to one transplant. There will be an inherent bias in the calculation of the EFS time. However, the longer duration is considered a clinical drawback, and so there will be no statistical adjustment in the EFS time for the extra treatment. The EFS analysis will be intent-to-treat starting from the time of randomization, i.e., just prior to the first transplant.
9.3.1.2 Endpoints for Additional Primary Objectives
The endpoint to determine benefit of topotecan addition is response at the end of induction therapy. The proportion of Responders (end of induction CR/VGPR) will be compared to an analogous cohort of Responders on A3973.
The endpoint for determination of local control will be the incidence rate of local recurrence (local recurrence events: progression or relapse at site of primary, to be determined by central review).
9.3.2 Non-compliance with randomization (i.e., CROSSOVERS):
A crossover is defined as a patient who does not receive the second myeloablative regimen, except for reasons of disease progression.
The majority of crossovers are anticipated to be patients randomized to receive two myeloablative consolidations, but only receive one. Very few crossovers, if any, will be patients who were randomized to receive one myeloablative consolidation but were treated with two. If patients cross over to the other treatment arm, the detectable difference between treatment groups will increase. If the detection of a difference is to be maintained, a greater number of events (i.e., larger sample size) would be needed in order to overcome the diluting effect (Table A). Updated power calculations have been performed assuming a crossover rate from AT1+AT2 to AT1; this update obviates the need for Tables A and B below.
Table A - Effect of the Proportion of Crossovers on the number of events and sample size required to maintain detection of 12% difference in the 3-year EFS rate
Detectable 3-yr EFS difference
Proportion of Crossovers
Inflation Factor*
Number of Events
Number of Eligible Stage 4 Randomized Pts
Accrual Duration (years)
12%
0
1
199
329
4.06
12%
0.05
1.09
211
359
4.43
12%
0.10
1.20
232
395
4.88
12%
0.15
1.33
256
438
5.41
12%
0.20
1.47
284
484
5.98
12%
0.25
1.63
318
536
6.62
12%
0.30
1.84
359
605
7.47
12%
0.35
2.09
409
688
8.49
extension of Piantadosi annual accrual rate of 81 patients per year
If we are unwilling to increase the sample size to compensate for the diluting effect of crossovers, the detectable EFS difference will increase (Table B).
Table B. - Detectable difference in 3-year EFS rate based on the proportion of crossovers
Proportion of Crossovers
Inflation Factor
Relative Risk*
3-year EFS rate (%) with two transplants
Detectable Difference in 2-yr EFS rate (%)
0
1
0.703
58.0
12.0
0.05
1.09
0.692
58.4
12.4
0.10
1.20
0.680
59.0
13.0
0.15
1.33
0.666
59.6
13.6
0.20
1.47
0.652
60.3
14.1
0.25
1.63
0.638
60.9
14.7
0.30
1.84
0.620
61.8
15.4
0.35
2.09
0.601
62.7
16.3
decreased risk for an event with two transplants
For the first 79 patients randomized to the two-transplant arm, the proportion of crossovers will be monitored using the two-stage rule below. Thereafter, it will be monitored using an upper bound of crossovers from AT1+AT2 to AT. If the proportion of crossovers from AT1+AT2 to AT reaches , then the detectable difference in an intent-to-treat analysis will have increased beyond a clinically
Children's Oncology Group
reasonable value of (i.e., beyond the expectation of increased benefit of two transplants). At that time, consideration should be given to modifying for increased accrual, modifying the study design, or abandoning the study due to infeasibility. Due to the dilution effect of the crossovers, an intent-to-treat comparison of treatment groups after 35% of patients have crossed from AT1+AT2 to AT would permit detection of a difference ( vs. ) in the 3 -year EFS rate. Stated another way, if of AT1+AT2 patients cross to AT, an underlying true difference of would need to exist in order for the study to be able to detect a difference of in an intent-to-treat analysis with . Per Table B , if the underlying true difference is only , and there are crossovers, then 688 patients would be required to detect the 12% difference.
9.3.3 Power for detection of difference in proportion of induction responders
All patients on this study will be compared to patients on A3973, for a total of 1,150 patients in the test of proportions. The power and detectable different were already very good per Table C, but with the increase in sample size from what was originally planned, the available power will be higher and/or the detectable difference will be smaller than what was originally planned (Table C). The A3973 cohort will be nearly identical to the cohort on this study with the exception of the change in the induction therapy under study and the later timeframe of this study. Comparison will be made to assess ability to achieve CR + VGPR.
Table C. - Detectable difference in CR + VGPR response rates with
Potential post-induction response rate on
A3973
Response rate on this study
Detectable
difference
Power
9.3.4 Power for detection of a difference in the risk of local recurrence
There are two differences between ANBL0532 and A3973 in the cohort of patients who have pretransplant <CR of the primary: 1) the number of transplants; and 2) the use of purging. For an unbiased comparison of analogous patients who achieve pre-transplant of the primary, the Treatment 01 patients on ANBL0532 ( 36 Gy EBRT) will be compared to the unpurged patients ( ) on A3973 (21.6 Gy EBRT) in terms of their risk for local recurrence. The following projections are based on response rates from prior protocols. We assume of unpurged patients on A3973, and ( ) of patients randomized to receive one transplant on this study will achieve less than CR at primary site at end of induction therapy. If of the A3973 patients and of the ANBL0532 patients make it from randomization to consolidation and XRT, then the total number of patients utilized for the local recurrence risk comparison is ( 83 from A3973 and 134 from ANBL0532). This will provide more than power to detect a clinically significant decrease of (from to ) in the risk for local recurrence after 36 Gy, and more than power to detect an decrease (from to ) in a one-sided Fisher's Exact test at a significance level of 0.05 .
9.3.5 Monitoring
a) Monitoring for the proportion of "crossovers" from the two-transplant arm to the one-transplant arm:
Of the eligible patients randomized to two transplants, patients who are unable to receive the second transplant except for reasons of disease progression ("crossovers") will be identified. The complement cohort who are compliant with their randomized assignment shall be called the "compliers". A two-stage rule will be used to monitor the proportion of "compliers" within the cohort of eligible patients randomized to receive two transplants.
Stage 1: Accrue 33 patients randomized to the two-transplant arm. If 20 or fewer patients are "compliers", then consider modifying the study design to maintain power. If more than 20 patients are "compliers", then proceed to Stage 2.
Stage 2: Accrue an additional 46 patients to the two-transplant arm for a total of 79 . If 53 or fewer patients are "compliers", then consider modifying the study design to maintain power.
This two-stage design has the following characteristics: The proportion of "compliers" is 0.61 under the null, and 0.75 under the alternative. This exact test will have power and a significance level of <0.10.
b) Interim monitoring for futility or superiority of the experimental arm (AT1-TC & AT2-CEM) will start after of the total expected information ( 177 events) has been observed, and will be performed on an annual basis. If issues of concern arise that require more frequent looks, then monitoring can be performed every six months.
There will be cause to stop the trial early if the AT1 + AT2 arm appears to be insufficiently efficacious or significantly more efficacious. The need for curtailment for non-significance of the AT1 + AT2 arm will be assessed using Fleming-Harrington-O'Brien boundaries for the hazard ratio (AT: AT1+AT2) at each interim analysis (Table D). The interim analyses of EFS will take place every year starting after of the planned events have occurred. The lower bound is calculated based on repeated testing of the alternative hypothesis that the relative risk is equal to 1.42 at a p-value of 0.005 . This relative risk is calculated as control: experimental using the planning parameters for 3-year EFS. The upper bound uses an alpha*t spending function for a cumulative alpha level of 0.05 .
Table D - Fleming-Harrington-O'Brien interim monitoring boundary values for log-rank EFS comparison of treatment arms
Timepoint from monitoring start (years)
Cumulative number of events expected
Proportion of Total Expected Information
Lower Boundary z-value
Upper Boundary z -value
0
36
0.201
-1.459
2.878
1
71
0.402
-0.996
2.470
2
102
0.578
-0.682
2.227
3
134
0.759
-0.406
2.033
4
158
0.889
-0.227
1.918
5
177
1.000
-0.085
1.840
c) Descriptive monitoring: In addition, the following descriptive monitoring will be performed. If descriptive results present a concern, then a formal statistical monitoring rule will be developed:
i) the proportion of local recurrences within the cohort of patients who have pretransplant <CR (about 25% expected);
ii) the induction toxic death rate (about expected);
iii) end of induction response rate (about for CR+VGPR);
iv) rate of PBSC contamination as determined by ICC (about expected) (this monitoring was discontinued starting with Amendment #2);
v) the toxic death rate post-consolidation attributed to TC or TC + CEM (about 5% expected); and,
vi) by ANBL0532 treatment arm, the proportion of patients who choose to enroll on ANBL0032 or ANBL0931 (difference less than 5%).
9.3.6 Assessment of Study Objectives
Primary Objective 1.1.1: Intent-to-treat log-rank test comparison of EFS curves, starting from the time of randomization, by treatment group (AT-CEM vs. AT1-TC & AT2-CEM). Kaplan-Meier curves will be generated starting from a) the time of randomization (this is the definitive analysis); b) the time of transplant; c) the time of completion of the last transplant (for descriptive purposes); and d) the time of diagnosis (for descriptive purposes).
Primary Objective 1.1.2: Chi-square test of proportions in all patients to compare the proportion of Responders (CR+VGPRs) at the end of induction in this study to the analogous proportion in A3973. There is no reason to believe that the characteristics of the induction cohort on this study will differ from that of the induction cohort on A3973, so this will be a fair and appropriate comparison.
Primary Objective 1.1.3: Log-rank test to compare the risk for local recurrence within the subset of patients who obtain a pre-transplant primary site CR, i.e., comparing ANBL0532 patients randomized to get one transplant versus the historical A3973 patients who were randomized to unpurged transplant.
Secondary Objective 1.2.1: The endpoints for this correlative aim are a) the duration of grade neutropenia during cycle one; b) the duration of grade thrombocytopenia during cycle one; and, c) the response rate after two cycles of induction therapy. Of the total 664 patients, we will assume that will have biologic samples submitted for this correlative study. For a) and b), a logistic regression model will be used to test the ability of the number of days of neutropenia or thrombocytopenia to predict the presence of a polymorphism. For c), a chi-square test of association will be used to compare the proportion of responders with versus without a polymorphism.
For a particular genetic polymorphism, if the prevalence of homozygocity is , the resulting sample sizes of 155 homozygous patients and 363 non-homozygous patients will result in more than power to detect a difference in response rates of versus , respectively, assuming a 2 -sided 0.05 alpha level. If the prevalence of homozygocity for a particular genetic polymorphism is , the resulting sample sizes of 259 homozygous patients and 259 non-homozygous patients will result in more than power to detect a difference in response rates of versus , respectively, assuming a 2 -sided 0.05 alpha level. If the prevalence of homozygocity for a particular genetic polymorphism is , the resulting sample sizes of 52 homozygous patients and 466 non-homozygous patients will result in more than power to detect a difference in response rates of versus , respectively, assuming a 2 sided 0.05 alpha level.
Secondary Objective 1.2.2: A chi-square test of proportions will be used to test for association between the proportion of patients who achieve a surgical CR and the proportion of patients who do not relapse in the primary. A logrank test will compare the EFS curves for degree of surgical response (CR vs. <CR).
Secondary Objective 1.2.3: The proportion of patients by type of surgical complication and type of radiation therapy complication will be descriptively tabulated. The complications, described in more detail in section 13.5.2, are: bowel obstruction, chylous leak, renal injury/atrophy/loss and diarrhea.
Secondary Objective 1.2.4: For the patients with primary tumors with intraspinal extension: descriptive tabulation of the proportion of patients with resolution of neurologic symptoms by type of symptom.
Secondary Objective 1.2.5: The cohort of patients for this analysis of the cis-RA PK data are patients enrolled on either A3973, ANBL0032, ANBL0931, ANBL0532, and future high risk studies who receive cisRA. The analyses will be conducted in the overall cohort as well as within the subset of patients who receive only one transplant and cis-RA without the addition of antibody. Patients will be pooled across these studies until at least 195 patients are available for analysis. We would not stop consenting patients to this aim or collecting blood specimens in the middle of a study simply because the minimum accrual goal of had
been reached; accrual of patients towards this aim would continue until that study's overall accrual was completed. A larger cohort would permit this aim to be addressed with greater accuracy.
1.2.5 a) For each patient, the peak serum concentration level will be determined. Pilot data indicate the mean peak serum concentration level will be approximately . Given a mean peak serum concentration level of , a sample size of will permit placement of a confidence interval on the mean peak serum concentration level with a precision of . This confidence interval will provide a descriptive assessment of the inter-patient variability of the cis-RA plasma levels.
To determine the relationship of peak serum concentration level with the existence of polymorphisms, Kendall's Tau statistic will be calculated. The allelic frequency of the known polymorphisms in UGT1A1 and UGT2B7 are such that the expected numbers of homozygotes for the variant forms are in caucasians. Therefore, collection of 80 patient samples would allow us to detect a 2 -fold difference in the peak serum concentration level of 13-cis retinoic acid with an expected power of greater than and a difference with a power of . The allelic frequency of the CYP2C8 variant, CYP2C83 in Caucasians is 0.13 , giving a frequency of only for homozygotes. However, the influence of this variant in those heterozygous for CYP2C83 has not been determined and evidence for variation between heterozygotes and those possessing the homozygous wild-type genotype exists for other genes. Combining heterozygotes and homozygous variants for this gene (CYP2C8) gives similar statistical power to that applied to the UGT polymorphisms.
1.2.5 b) To determine if there is a relationship of the peak serum concentration level with event-free survival, the term for this level will be tested in a Cox proportional hazards model. If the level is statistically significant, then a) the hazard ratio will provide a measure of increased risk for an event for a unit change in the level; and, b) a cut-off in serum concentration level (low vs. high) will be identified that optimizes the difference in EFS between the low and high serum concentration groups.
To determine if there is a relationship of the peak serum concentration level with toxicity rates (CTC grade 3 or 4 skin, hypercalcemia, or hepatic toxicity), Kendall's Tau statistic will be calculated.
1.2.5 c) To determine if pharmacogenomic variations are predictive of EFS, a logrank test comparison of patients with vs without a given polymorphism will be made.
A Fisher's exact test will test for association of the presence of a polymorphism with the occurrence of systemic toxicity (CTC grade 3 or 4 skin, hypercalcemia, or hepatic toxicity).
These tests will be performed for UGT1A1, UGT2B7, CYP2C8 and CYP3A7 alleles.
Secondary Objective 1.2.6: The hypothesis for this aim is that topotecan systemic clearance can be calculated with acceptable accuracy and precision using patient specific covariates in a dosing model. The topotecan total clearance value will be calculated from a single sample using Bayesian modeling techniques and population priors established from a similar patient population studied by Dr. Stewart's laboratory. The two clearance values from course 1 and 2 will be used in the analysis separately and as an average value. In addition to this topotecan clearance, a "calculated" topotecan clearance value will be determined from a topotecan dosing model, which consists of patient specific covariates determined from a) previous population pharmacokinetic analyses, b) planned population pharmacokinetic analyses of already collected data, and c) population pharmacokinetic analyses of data that will be collected during this clinical trial. The relation between the topotecan clearance value determined by pharmacokinetic analysis (limited sampling; Bayesian modeling) and the topotecan clearance value determined by the topotecan dosing model (patient covariates) will be investigated using regression analysis and simulation studies. The long-term goal is to identify relevant patient covariates that will enable the development of a
clinically useful dosing nomogram for topotecan in children with cancer. This approach will minimize the interpatient variability in topotecan systemic exposure by individualizing the topotecan dosage.
Secondary Objective 1.2.7. We hypothesize that: i) cells recognizing antigens expressed on neuroblastoma cells circulate at diagnosis, and ii) anti-neuroblastoma T cells can demonstrate effector function against neuroblastoma cells, We will assess functional immunity against neuroblastoma in patient samples, including T cells collected at diagnosis and in patients after myeloablative therapy. We will look for functional T cell responses using both a specific tumor antigen (survivin) as well as allogeneic tumor RNA as a source of antigenic stimulus.
Assays used will include:
Survivin-specific cytotoxic T lymphocytes detected using peptide/MHC tetramers in HLA-A2+ patients. This assay provides measures of the presence of T-cells that are capable of recognizing neuroblastoma. For objective 1.2.7.a, the median number of survivin-specific T-cells will be calculated at diagnosis and post-transplant. The proportion of patients who are positive for survivin-specific immunity will be calculated at diagnosis and post-transplant. For objective 1.2.7.d, of the patients who were positive for survivin-specific immunity at diagnosis, the proportion of patients who remain positive post-transplant will be calculated.
IFN-gamma production in ELISPOT assays to antigen presenting cells (APCs) loaded with tumor RNA, survivin RNA, or control RNA. This assay provides a measure of the number of T-cells that are capable of recognizing neuroblastoma. A sample of peripheral blood mononuclear cells (PBMC) will be exposed to
APCs loaded with mRNA for survivin, mRNA from neuroblastoma cells and irrelevant mRNA. This assay does not require that the patient be HLA-A2+. For objective 1.2.7.b, the median number of T-cells and the median number of IFN-gamma-producing T-cells will be calculated using T-cells collected at diagnosis and after completion of myeloablative therapy for each stimulating APC type. For each stimulating APC type, the median number of T-cells after transplant will be descriptively compared to a) the median number of T-cells at diagnosis; and, b) the median number of IFN-gamma-producing T-cells at diagnosis.
3. Response of APC-stimulated cytotoxic T-lymphocytes (CTL) response to neuroblastoma cells. The ability to lyse neuroblastoma cells will be evaluated for T cells stimulated by exposure to APCs loaded with mRNA for survivin, mRNA from neuroblastoma cells and irrelevant mRNA. The ability of stimulated T-cells to lyse neuroblastoma cells will be calculated at diagnosis and after myeloablative therapy. This assay provides a measure of the ability of stimulated T-cells to kill neuroblastoma cells. For objective 1.2.7.c, the proportion of patients whose stimulated T-cells lyse neuroblastoma cells will be descriptively compared between diagnosis and after completion of myeloablative therapy.
It is not necessary to perform treatment arm comparisons to assess this objective.
Secondary Objective 1.2.8. We hypothesize that patients who received the tandem myeloablative chemotherapy regimen will experience slower T cell recovery, specifically of CD4+ T cells when compared to patients who received a single myeloablative chemotherapy regimen. Enumeration of peripheral blood CD3, CD4 and CD8 cells will be performed at 1,3 and 6 months after completion of assigned myeloablative therapy. For each timepoint ( months post-myeloablative therapy), we will perform a descriptive comparison of the median number of T-cells (CD3, CD4, CD8) between treatment arms (single vs. tandem myeloablative regimens).
Children's Oncology Group
Secondary Objective 1.2.9 - The proportion of patients with neuroblastoma detected in bone marrow and peripheral blood using RT-PCR technique will be calculated overall and by treatment arm. Data from CCG-3891 indicate that by immunocytology of patients prior to myeloablative therapy show minimal residual disease in bone marrow, and of patients in peripheral blood at the time of bone marrow harvest. Presumably residual disease found after transplant will be less than this figure, although the sensitivity of RT-PCR may increase the proportion of neuroblastoma positive specimens. If of the randomized patients are positive for neuroblastoma by bone marrow RT-PCR or immunocytochemistry and if the EFS from time of transplant of the positive group is two thirds that of the negative group, this study will have good power ( ) to detect this difference. The same considerations apply to peripheral blood; unless the EFS difference is quite large between the groups this study will have limited power to detect differences by peripheral blood positivity. The statistical tool used will be the log rank test, stratified by any other variables of interest. Cox regression techniques will be used for multivariate analysis, in particular for determination of whether these variables add independent prognostic information.
Secondary Objective 1.2.10
A descriptive analysis of outcome will be performed of patients months with Stage 4, MYCN nonamplified tumor with unfavorable histopathology or diploid DNA content or with indeterminant histology or ploidy and patients who are greater than 547 days of age with Stage 3, MYCN nonamplified tumor AND unfavorable histopathology or indeterminant histology. Kaplan-Meier curves of EFS and OS will be plotted, and the proportion of responders to induction therapy will be tabulated.
9.4 Gender and Minority Accrual Estimates
The gender and minority distribution of the study population is expected to be:
Accrual Targets
Ethnic Category
Sex/Gender
Females
Males
Total
Hispanic or Latino
27
35
62
Not Hispanic or Latino
257
345
602
Ethnic Category: Total of all subjects
284
380
*664
Racial Category
American Indian or Alaskan Native
1
1
2
Asian
10
21
31
Black or African American
37
54
91
Native Hawaiian or other Pacific Islander
1
1
2
White
235
303
538
Racial Category: Total of all subjects
284
380
*664
These totals must agree
This distribution was derived from A3973 .
10.0 EVALUATION CRITERIA
10.1 Common Terminology Criteria for Adverse Events (CTCAE)
This study will utilize the CTCAE of the National Cancer Institute (NCI) for toxicity and performance reporting. The descriptions and grading scales found in the revised CTCAE version 4.0 will be utilized for reporting beginning July , 2011. All appropriate treatment areas should have access to a copy of the CTCAE version 4.0 , which can be downloaded from the CTEP web site (http://ctep.cancer.gov).
10.2 Response Criteria for Patients with Solid Tumors
This study will use the International Neuroblastoma Staging System (INSS) Response Evaluation Criteria. This will allow direct comparison with legacy COG studies as well as Phase 3 trials performed abroad.
10.3 International Staging System
Stage 1: Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative microscopically (nodes attached to and removed with the primary tumor may be positive).
Stage 2A: Localized tumor with incomplete gross excision; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically.
Stage 2B: Localized tumor with or without complete gross excision; with ipsilateral nonadherent lymph nodes positive for tumor. Enlarged contralateral lymph nodes must be negative microscopically.
Stage 3: Unresectable unilateral tumor infiltrating across the midline with or without regional lymph node involvement; or, localized unilateral tumor with contralateral regional lymph node involvement; or, midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement. The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column.
Stage 4: Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin and/or other organs (except as defined for Stage 4S).
Stage 4S: Localized primary tumor (as defined for Stage 1 or 2A or 2B), with dissemination limited to liver, skin, and/or bone marrow (limited to infants year of age). Marrow involvement should be of total nucleated cells identified as malignant on bone marrow biopsy or on marrow aspirate. MIBG scan (if performed) should be negative in the marrow.
10.4 International Response Criteria
Measurable tumor is defined as the product of the longest x widest perpendicular diameter. The third dimension is added when possible. Elevated catecholamine metabolite levels and tumor cell invasion of bone marrow also is considered measurable tumor. See Appendix VI.
10.4.1 Complete Response (CR)**
No evidence of primary tumor, no evidence of metastases (chest, abdomen, liver, bone, bone marrow, nodes, etc.), and HVA/VMA normal.
10.4.2 Very Good Partial Response (VGPR)
Greater than reduction of primary tumor; no metastatic tumor (as above except bone); no new bone lesions, all pre-existing lesions improved on bone scan; HVA/VMA normal.
ANBL0532
10.4.3 Partial Response (PR)
Fifty-90% reduction of primary tumor; 50% or greater reduction in measurable sites of metastases; 0-1 bone marrow samples with tumor; number of positive bone sites decreased by .
10.4.4 Mixed Response (MR)
Greater than 50% reduction of any measurable lesion (primary or metastases) with, <50% reduction in any other site; no new lesions; <25% increase in any existing lesion (exclude bone marrow evaluation).
10.4.5 No Response (NR)
No new lesions; <50% reduction but <25% increase in any existing lesion (exclude bone marrow evaluation).
10.4.6 Progressive disease (PD)**
Any new lesion or increase of a measurable lesion by ; previous negative marrow positive for tumor.
** MIBG scan must be negative for patient to be classified as having a complete response. New site of disease documented by MIBG scan qualifies patient as having progressive disease.
11.0 ADVERSE EVENT REPORTING REQUIREMENTS
11.1 Purpose
Adverse event data collection and reporting, which are required as part of every clinical trial, are done to ensure the safety of patients enrolled in the studies as well as those who will enroll in future studies using similar agents.
11.2 Determination of Reporting Requirements
Reporting requirements may include the following considerations: 1) the characteristics of the adverse event including the grade (severity); 2) the relationship to the study therapy (attribution); and 3) the prior experience (expectedness) of the adverse event. The CTCAE (v 4.0 will be used starting from July , 2011) provides descriptive terminology and a grading scale for each adverse event listed.
Commercial agents are those agents not provided under an IND but obtained instead from a commercial source. In some cases an agent obtained commercially may be used for indications not included in the package label. In addition, NCI may on some occasions distribute commercial supplies for a trial. Even in these cases, the agent is still considered to be a commercial agent and the procedures described below should be followed.
Determine the prior experience Expected events are those that have been previously identified as resulting from administration of the agent. An adverse event is considered unexpected, for reporting purposes only, when either the type of event or the severity of the event is not listed in:
the current NCI Agent-Specific Adverse Event List (provided in the Drug Information Section of this protocol); or
the drug package insert (for treatments with commercially available agents).
11.3 Reporting of Adverse Events for Commercial Agents - AdEERS abbreviated pathway
Commercial reporting requirements are provided in Table B. The commercial agent(s) used in this study are listed in the Drug Information Section of this protocol.
ANBL0532
COG requires the AdEERS report to be submitted within 5 calendar days of learning of the event. Use the NCI protocol number and the protocol-specific patient ID provided during trial registration on all reports.
Table B
Reporting requirements for adverse events experienced by patients on study who have NOT received any doses of an investigational agent on this study.
AdEERS Reporting Requirements for Adverse Events That Occur During Therapy With a Commercial Agent or Within 30 Days
Attribution
Grade 4
Grade 5
Unexpected
Expected
Unrelated or Unlikely
AdEERS
Possible, Probable, Definite
AdEERS
AdEERS
This includes all deaths within 30 days of the last dose of treatment with a commercial agent, regardless of attribution. Any death that occurs more than 30 days after the last dose of treatment with a commercial agent which can be attributed (possibly, probably, or definitely) to the agent and is not due to cancer recurrence must be reported via AdEERS.
11.4 Routine Adverse Event Reporting
The NCI defines both routine and expedited AE reporting. Routine reporting is accomplished via the Adverse Event (AE) Case Report Form (CRF) within the study database. For this study, routine reporting will include all AdEERS reportable events and Grade 4 and higher non-hematologic Adverse Events with any attribution. Reporting of hematologic adverse events will not be required as a part of routine reporting for this study. Exceptions to the routine reporting criteria above are the following which are necessary to meet study aims:
Grade 3 or higher neutropenia and/or thrombocytopenia during induction Cycle 1
Grade 2 or higher diarrhea or gastrointestinal obstruction which may occur at any time after surgical resection of primary tumor.
11.5 Reporting Secondary AML/MDS
As of August 25, 2010, all secondary malignancies should be reported via AdEERS.
12.0 RECORDS AND REPORTING
12.1 Categories of Research Records
Research records for this study can be divided into three categories:
Non-computerized Information: Pathology Narrative Reports and Surgical Reports. These forms are submitted through the Document Imaging System in the eRDES
Children's Oncology Group
2. Reference Labs' required reports, and QARC data: These data accompany submissions to these centers, which forward their review data electronically to the COG Statistics and Data Center.
3. Computerized Information Electronically Submitted: All other computerized data will be entered in the COG Remote Data Entry System with the aid of schedules and worksheets (essentially paper copies of the RDE screens) provided in the data form packet.
See separate Data Form Packet posted on the COG web site, which includes submission schedule.
12.2 CDUS
This study will be monitored by the Clinical Data Update System (CDUS). Cumulative CDUS data will be submitted quarterly to CTEP by electronic means. Reports are due January 31, April 30, July 31 and October 31. This is not a responsibility of institutions participating in this trial.
13.0 SURGICAL GUIDELINES
13.1 Surgical rationale
The overall surgical goal in high-risk patients with neuroblastoma is the most complete tumor resection with preservation of full organ and neurologic function if possible. In addition, the surgeon is responsible for the preservation and delivery of an adequate surgical specimen to the appropriate laboratory for crucial biologic analyses; including determination of MYCN amplification, ploidy, and molecular genetic analyses. Titanium clips should be placed around sites of residual disease.
13.2 Pre-operative management
Adequate pre-operative imaging of the primary tumor and sites of regional spread is done by either computerized axial tomography, magnetic resonance imaging, or a combination of the modalities. Sites of distant metastases should be evaluated by a combination of clinical and bone marrow assessment as well as bone scan and MIBG scans. When dealing with paraspinal or epidural lesions pre-operative neurosurgical consultation is recommended and a baseline neurologic assessment carried out (see section 13.4.3 and Appendix III). The planned operation should be discussed with the attending pediatric oncologist and, if possible, at tumor board and the goals of the surgery should be clearly understood by all involved services pre-operatively.
13.3 Sampling requirements
In all patients, the primary purpose of the initial surgical procedure is to obtain enough tissue to establish the diagnosis, determine stage, and secure enough properly preserved tumor for biological studies. Refer to ANBL00B1 for complete details of specimen requirements. All patients must be enrolled onto ANBL00B1 to be eligible to participate in ANBL0532. An adequate biopsy to determine the diagnosis and assess biological variables like histopathologic classification, MYCN amplification and ploidy is required. Usually more than 1 cubic centimeter of viable tissue is needed for all these assays. Needle biopsies are not sufficient for histologic classification. The surgeon should remember that a significant portion of the tumor may be necrotic. Frozen section examination of a small amount of tissue will verify that viable neuroblastoma is being biopsied. The anesthesia time while awaiting frozen section analysis can also be used to obtain bone marrow aspirations and biopsies, and for placement of a vascular access device.
For patients with stage 4 S disease who are very ill and in whom an open biopsy to obtain tissue for diagnosis and biologic studies is considered medically contraindicated, every effort should be made to obtain some tumor tissue by fine needle aspiration of a metastatic site for at minimum the determination of status.
ANBL0532
13.4 Operative management
13.4.1 Central Line Placement
Patients will require central venous access both for treatment and apheresis. It is usually feasible and efficient to place a vascular access device and obtain a bone marrow aspirate and biopsy during the same anesthetic. The appropriate catheter should be placed from initiation of therapy.
Medcomp or similar catheters are specifically designed as tunneled, permanent apheresis catheters. It may not be possible to draw at a sufficient rate from a non-apheresis catheter that is smaller than 10Fr. If a smaller double lumen must be placed, or if it is not possible to draw at a sufficient rate ( ) then it may be necessary to place an additional single lumen catheter for the apheresis. If a second, single-lumen line is needed, it is best to place it electively PRIOR to starting the mobilization cycle of chemotherapy (i.e., Cycle 2 induction chemotherapy).
13.4.2 Diagnostic Surgery
In all patients, the primary purpose of the initial surgical procedure is to obtain enough tissue to establish the diagnosis, determine stage, and secure enough properly preserved tumor for biological studies. The great majority of high-risk patients will undergo initial diagnostic biopsy without resection. Biopsy of the primary tumor or an accessible metastatic site is acceptable. The goal of the biopsy procedure is to obtain enough tissue for a histopathological diagnosis as well as MYCN determination, cytogenetics, and other biological studies. The surgeon should try to obtain at least one of viable tumor tissue, if feasible, according to the surgeon's judgment. Complete excision of the primary tumor can occasionally be performed if the tumor is easily resectable without a lengthy procedure or extensive dissection. However, a resection should not be undertaken if it might result in significant delay in the initiation of chemotherapy or great morbidity. In some institutions the diagnosis is established by finding neuroblastoma in bone marrow specimens in conjunction with elevated urinary catecholamines.
13.4.3 Tumors with Intraspinal Extension
When the tumor approaches the spinal canal on imaging (see Section 16.2), a detailed examination must assess neurological function. The format for the examination, a modification of the ASCIA scale, is detailed in Appendix III and should be used to document the degree of neurologic dysfunction at diagnosis and at subsequent time points during and post-therapy.
Laminectomy should not be performed in patients who are neurologically asymptomatic. Patients with symptomatic spinal cord compression secondary to intraspinal/epidural extension of neuroblastoma through a neural foramina may require laminectomy, or osteoplastic laminotomy at diagnosis to prevent permanent paralysis. However, treatment with chemotherapy alone or chemotherapy and radiation therapy will frequently be sufficient to rapidly reverse symptoms of cord compression. Therapeutic decisions in neuroblastoma patients with spinal cord compression should be made with the multidisciplinary involvement of the attending pediatric oncologist, general pediatric surgeon, and pediatric neurosurgeon.
If neurologic deterioration occurs during chemotherapy, neurosurgical evaluation should be sought and operative decompression strongly considered. Appropriate to the degree of neurological impairment, the treating physicians may decide that operative neurosurgical decompression is indicated under these circumstances. If feasible, the neurosurgeon should perform an osteoplastic laminotomy, with secure replacement of the laminae after decompression has been accomplished. Operative details will be recorded in the RDE.
Assessment of neurological function will be performed prior to start of treatment, after 2 cycles of inductions, end-induction therapy, end consolidation therapy and end of cisRA therapy for all patients
with intraspinal extension of tumors (see Section 7.0). Neurologic and orthopedic evaluations will be a routine part of off therapy follow-up for patients with intraspinal extension of tumors, and will be performed at , and 36 months post therapy, and then annually thereafter. Orthopedic evaluation will include assessment of scoliosis and of extremity deformity by upright spine x-ray at these time points (see Appendix III).
13.4.4 Operative management of primary tumors after chemotherapy
The majority of patients will undergo resection of the primary tumor after initial induction chemotherapy. Surgical resection should be performed when the and the patient is medically stable after Cycle 5 of Induction. This is to allow maximal tumor reduction by chemotherapy prior to resection and to reduce vascularity. Surgical resection may be performed later in Induction, if necessary, but must occur prior to Consolidation. Surgical scheduling SHOULD AVOID DELAYS OF MORE THAN SIX WEEKS BETWEEN CHEMOTHERAPY CYCLES.
The goal of delayed surgery is gross total resection of residual tumor in the primary site as well as tumor in areas of regional dissemination (usually lymph nodes). Resection with microscopically negative margins may not be feasible because of proximity to major vascular structures and the spine. Instead, the surgeon should concentrate on removing, as completely as possible, all gross disease. It is acceptable, and often necessary, to incise the tumor and remove it in a segmental fashion. Titianium clips should be used to mark all areas of residual disease. All attempts should be made to preserve organs, especially the kidney. Rarely, nephrectomy may be necessary for complete tumor removal but this should only be planned if the involved kidney has greatly diminished function. If pre-operatively a nephrectomy is being considered, then a differential GFR should be obtained to determine what the renal function will be in the remaining kidney. This is extremely important because carboplatin is used for consolidation and toxicity may be greater for patients with a GFR . The surgical committee of the Children's Oncology Group strongly recommends kidney preservation when feasible.
It is vital that the operating surgeon dictate a detailed operative note, which should include: the completeness of resection, areas of residual disease, estimated blood loss, and any operative complication such as identification of injury to adjacent structures, removal of normal organs, renal injury and vascular injury.
Surgical resection of residual disease following HSCT is not encouraged, unless it would alter therapy.
13.5 Management of Surgical Complications
13.5.1 Intraoperative Complications
Intraoperative complications are site-dependent. Major hemorrhage from either venous or arterial structures is always possible with these infiltrative tumors. The principles of vascular surgery, including proximal and distal control, pertain. Appropriate intraoperative vascular consultation should be sought if necessary. Crucial vessels like the carotid, subclavian, hepatic, superior mesenteric or renal arteries should be repaired and flow restored even if bypass grafting is required. Nerve injuries may also be incurred and should be primarily repaired using magnification.
13.5.2 Post-operative Complications
This topic is too broad for simple discussion. Generally, large neuroblastoma resections result in significant third space losses and require vigorous fluid replacement. Because of this fluid requirement patients may require significant periods of post-operative ventilation. The need for post-operative monitoring in an intensive care environment should be anticipated. Acute and long-term complications and duration of complications will be prospectively monitored, including chylous leak (thoracic and
abdominal); secretory non-infectious diarrhea; bowel obstruction due to post-surgical adhesions and renal dysfunction due to vascular injury.
Pulmonary Complications: Resection of large abdominal neuroblastoma may result in significant third space losses and require vigorous fluid replacement intraoperatively and postoperatively. Because of this fluid requirement patients may require significant periods of post-operative ventilation. The need for post-operative monitoring in an intensive care environment should be anticipated. Pneumonias or other pulmonary complications related to ICU and postoperative course (including incubation days) should be reported
Bowel Obstruction: Small bowel obstruction may occur for many reasons, including injury, exposure, or blunt trauma during the resection, postoperative intussusception, formation of adhesions, or related to radiation injury. Obstruction may occur in the early postoperative period, or may occur months to years later. This study will collect long term data on the occurrence, etiology, and management of small bowel obstruction in this patient population, with the aims of reporting accurate occurrence rates, along with correlation of occurrence to the extent of resection and other efforts at local control.
Chylous Leak: Extensive dissection of the retroperitoneum or mediastinum may result in either frank disruption of chylous channels, or interruption causing intraluminal lymphatic hypertension and leak. This study will collect long term data on the occurrence and evaluation of abdominal distension, chylous ascites, and chylothorax as they relate to chylous leak, with the aims of reporting accurate occurrence rates, along with correlation of occurrence to extent of resection and other attempts at local control
Renal Injury/Atrophy: Renal injury may manifest in many forms, related to the nature of the injury or insult. While nephrectomy is strongly discouraged, extensive perirenal dissection and kidney sparing surgery may still result in vascular occlusion, infarction, traumatic compression, or other injury mechanisms sufficient to cause long term dysfunction or atrophy. Additionally, radiation therapy may lead to kidney damage, again with the long term finding of atrophy. This study will collect long term data on renal function, identifying the occurrence of renal injury and atrophy, with the aims of reporting accurate occurrence rates, along with correlation to extent of resection and other attempts at local control.
Diarrhea: Extensive sympathetic denervation associated with aggressive retroperitoneal dissection may result in increased frequency of stooling or diarrhea. This study will collect long term data on the occurrence and interventions necessary for the control of postoperative diarrhea, with the aims of reporting accurate occurrence rates, along with correlation to extent of resection and other attempts at local control.
13.6 Special techniques
13.6.1 Nerve Stimulation
Nerve stimulation can be useful in detecting motor nerves in the brachial or lumbo-sacral plexus. This requires cooperation from the anesthesiologist as muscle relaxation must be allowed to wear off. Nerve stimulation should always be used when dissection along the pelvic sidewall or in the neck or thoracic inlet.
13.6.2 Ultrasonic Dissector-aspirators
Some authors have described the use of ultrasonic dissectors (CUSA) to debulk the interior of large tumors allowing an easier capsular dissection. The technique is useful for friable tumors but not those that are stroma rich. The surgeon should try to perform a generous incisional biopsy prior to ultrasonic dissection as it is difficult to capture the tumor specimen after it has been aspirated into the device.
ANBL0532
13.6.3 Thoracoscopy
Video-assisted thoracoscopy can be used to remove small posterior mediastinal or thoracic inlet tumors provided there is no vascular encasement. One-lung ventilation is mandatory.
13.6.4 Laparoscopy
Laparoscopic resection of small adrenal or pelvic primaries can be done. Extensive tumors or those with significant vascular encasement, or locoregional nodal spread can be more completely resected using standard open approaches.
13.6.5 Radiofrequency Ablation, Cryosurgery
These techniques have significant drawbacks when applied to lesions in proximity to major vascular structures and should be avoided when treating neuroblastoma.
13.7 Specimen requirements
As noted above an initial biopsy should obtain greater than 1 cubic centimeter of viable tumor tissue. Placement of the specimen in formalin should be avoided. Rather, the pathologist should be alerted and the specimen rapidly transferred from the operating room fresh and sterile. The surgeon should verify with the pathologist that viable tissue was sent and is being processed for COG biological studies and histopathology. Since it is hoped to extract RNA from these specimens, rapid freezing of some of the tissue is required.
14.0 PATHOLOGY GUIDELINES AND SPECIMEN REQUIREMENTS
14.1 Rapid Pathology Review
A rapid review of histology (under ANBL00B1) is required to determine eligibility of patients for this study. See COG ANBL00B1 for complete details on specimen requirements. Recently, as part of the international cooperative effort to develop a complete set of International Neuroblastoma Risk Groups, the International Neuroblastoma Pathology Committee devised a morphologic classification of neuroblastoma tumors that is modeled on the one proposed by Shimada and colleagues In ANBL00B1, tumor sections will be centrally reviewed, and classified as favorable or unfavorable according to the criteria described by the International Neuroblastoma Pathology Committee (INPC).
Send representative slides to the COG Pathology Center via Federal Express for overnight delivery. The slides will then be forwarded to Dr Hiroyuki Shimada, who will determine the histologic classification (INPC) and will notify the Neuroblastoma Biology Tracking Center. The Tracking Center will then determine protocol assignment (low, intermediate, and high-risk) and will notify the treating institution of the results.
14.1.1 Tracking Center Activity
Institutions will first enroll patients on ANBL00B1 once the patient's age, stage of disease and diagnosis have been determined. The COG Statistical Office will notify the Tracking Center with the enrollment information. Results of and ploidy status will be sent to the Tracking Center and to the treating institution by the Neuroblastoma Biology Reference Laboratory as soon as the results are available. The Tracking Center will notify immediately Dr Shimada in the event that a patient's stage, age, , and ploidy status mandate immediate review of histology for risk-grouping and treatment assignment. The Tracking Center will then notify the Statistical Office and the treating institution of the patient's risk group and protocol assignment. Patients will then be enrolled on the appropriate therapeutic study.
14.1.2 Diagnostic Concordance Study
In order to perform a concordance study between local institution and central review determination of the INPC classification, the Neuroblastoma Pathology Checklist is required to be completed by the institutional pathologist when tumor tissue is obtained at diagnosis.
14.1.3 Review Material Required (See ANBL00B1 for complete submission details)
Review materials are submitted through ANBL00B1. Do not submit separate pathology review materials for each protocol. Required for submission:
a. H&E stained slides from each paraffin block.
b. Representative paraffin blocks are preferred. If blocks are not available and slides are sent, please send 10 unstained sections from the most representative block(s) on coated slides for immunohistochemistry use
c. Copies of institutional Operative and Pathology reports
d. Copy of electron microscopy report, if available
e. Copy of Neuroblastoma Pathology checklist completed by the institutional pathologist
f. COG Neuroblastoma Biology Data Sheet and Shipping Form
Label pathology review materials with the COG patient ID number and the Surgical Pathology ID (SPID) number from the corresponding pathology report. Ship by Federal Express Priority Overnight (2504-6481-9) to the Biopathology Center.
14.1.4 Review Material at Second Look or Definitive Surgery and/or Relapse
Pathology materials must be submitted at the time of second look surgery, whether this procedure is a biopsy, partial resection, or complete resection, and/also at the time of relapse (see ANBL00B1 for fresh specimen requirements).
At subsequent surgeries and at relapse send:
a. 2 H&E and 6 unstained slides of residual tumor, both primary and metastatic, or send paraffin blocks.
b. Copies of institutional Operative and Pathology reports
c. Copy of electron microscopy report, if available
d. Copy of Neuroblastoma Pathology checklist completed by the institutional pathologist
e. COG Neuroblastoma Biology Data Sheet and Shipping Form
Label pathology review materials with the COG patient ID number and the Surgical Pathology ID (SPID) number from the corresponding pathology report. Ship to the Biopathology Center by regular mail or using your institution's courier account.
14.1.5 Autopsy Review
If autopsy permission is obtained, please send the following material:
a H&E sections of each major organ or paraffin blocks
b 2 H&E and 6 unstained slides of residual tumor, both primary and metastatic, or send paraffin blocks.
c Copy of the autopsy report.
d COG Neuroblastoma Biology Data Sheet and Shipping Form
Label the autopsy materials with the COG patient ID number and the Surgical Pathology ID (SPID) number from the corresponding pathology report. Ship to the Biopathology Center by regular mail or using your institution's courier account.
Children's Oncology Group
14.2 Specimen Shipping
Submit materials to:
Study Pathologist of record:
Hiroyuki Shimada, M.D.
Department of Pathology
Children's Hospital Los Angeles
4650 Sunset Blvd
Los Angeles, CA 90027
Phone: (323) 361-2377
Fax: (323) 667-1123
Email: hshimada@chla.usc.edu
15.0 SPECIAL STUDIES SPECIMEN REQUIREMENTS
15.1 Biologic Requirements
Patients must be enrolled on ANBL00B1 prior to the time of enrollment on ANBL0532. Enrollment on ANBL00B1 is required for all newly diagnosed patients within 21 days of diagnosis for all patients, with the exception of infants with stage 4 S neuroblastoma who are too ill to undergo a diagnostic biopsy procedure. Tissue procurement is mandatory for biology study registration. Consent for ANBL00B1 must be obtained at the time of tissue submission and should be within one week of surgery. Needle biopsies are not sufficient for histologic classification. Investigators are strongly encouraged to obtain adequate tissue (see sections 13.0 and 14.0) via open biopsy techniques. See protocol ANBL00B1 for details on the type of specimen to be obtained, how the specimen should be prepared, when the specimen should be shipped and the appropriate contact information.
ANBL00B1 enrollment allows tumor tissue and blood samples obtained at the time of diagnosis to be tested for the determination of INPC histologic classification, MYCN copy number and tumor cell ploidy. These results are used in real time to determine risk categorization and treatment assignment, especially for patients age 12-18 months with stage 4 disease or stage 3 disease. Residual tissues may be used for additional research testing and/or banking if informed consent for these options are indicated on the patient's ANBL00B1 consent document.
Study enrollment on ANBL0532 must be within 4 weeks of diagnosis, with confirmed biology status for initial risk group assignment.
15.2 Cyclophosphamide Pharmacogenomics
Patients may opt to participate in the pharmacogenomics study of cyclophosphamide. For these studies, peripheral blood should be drawn prior to starting therapy. The sample is, however, acceptable at any time the patient is not neutropenic.
Handling
Blood samples ( 10 mL ) are to be collected in a purple top tube (EDTA). Please label tubes with date of collection and patient COG identification number. The samples should be kept at room temperature.
ANBL0532
Shipping
The samples should be packaged to prevent damage and may be shipped at room temperature. Samples should be sent by Federal Express OVERNIGHT DELIVERY (2504-6481-9) for delivery only on Monday through Friday (samples obtained on Friday should be shipped on Monday by OVERNIGHT DELIVERY).
Send Samples to:
Linda Risler
Shen/McCune Lab
UW Dept of Environmental Health
4225 Roosevelt Way NE Suite 100
Seattle WA 98105.
Phone prior to shipping: 206-685-1650
Fax: 206-543-3835
15.3 Topotecan Pharmacokinetics
Specimen collection for this test is optional depending on whether or not the patient has signed an informed consent for them.
Topotecan Pharmacokinetic Studies Methods:
Samples for topotecan pharmacokinetics will be obtained from consenting patients minutes after the end of the topotecan infusion on Day 1 of Cycles 1 and 2.
Sample Timing, Processing, and Shipping
At least two weekdays prior to enrolling a patient on study, contact the Stewart Lab (St Jude Children's Research Hospital) at (901) 595-2400 between 8:00 am and 5:00 pm Central Time to request a Sample Collection Kit. The Sample Collection Kit will contain all paperwork and materials needed for sample collection, processing, and shipping. If chemotherapy must be started prior to arrival of kit, contact the Stewart Lab to obtain instructions for sample acquisition.
Sample Timing
On Day 1 of Cycles 1 and 2, obtain 2 mL of whole blood minutes after the end of the topotecan infusion. Please draw blood from a lumen other than the one from which topotecan was infused. Record the start and end time the topotecan infusion, along with the date and time of sample collection on the Pharmacokinetics Data Collection Form.
Instructions for Sample Processing and Shipping:
Place the whole blood specimen in a green-top heparin tube (provided), and mix by inversion five times. Record the exact time of day that the sample is obtained along with the time of day that the drug is administered on the Pharmacokinetics Data Collection Form provided in the kit.
Divide the whole blood sample equally into two properly labeled screw top tubes (provided) by using the transfer pipette provided in the pharmacokinetics kit.
Immediately place the whole blood samples on wet ice and store at within 15 minutes of collection.
Document on the Pharmacokinetic Data Collection Form all concomitant medications that the patient has taken within 48 hours of the pharmacokinetic study, including doses and dates started/stopped, especially dexamethasone and sulfamethoxazole/trimethoprim (Bactrim or Septra®).
The whole blood samples may be stored in a freezer until shipped. "Batching" of multiple patient samples must be approved by Dr. Clinton Stewart or his designee.
When ready to ship, place all vials in a plastic bag. Place the Pharmacokinetics Data Collection Form in a separate plastic bag. Place samples and five pounds of dry ice in the box provided along with paper towels or other absorbent packing material.
Whole blood samples and the Pharmacokinetic Data Collection Form should be sent by FEDEX OVERNIGHT DELIVERY for delivery Tuesday through Friday only. Samples obtained on Friday should be refrigerated until they can be shipped on Monday by OVERNIGHT DELIVERY.
Use the postage paid Federal Express shipping slip provided, which is already addressed to St. Jude. Before shipment contact the CRA, at (901) 595-5113. Call Federal Express at 1-800-238-5355.
NOTE: Use the Federal Express airbill included in the Sample Collection Kit for shipping samples at NO CHARGE.
Shipping Address
Stewart Laboratory
Pharmaceutical Sciences Department,
Chili's Care Center I5500
St. Jude Children's Research Hospital
262 Danny Thomas Place
Memphis, TN 38105
Tel. (901) 595-2400
Fax (901) 525-6869
15.4 Minimal Residual Disease Studies - Bone Marrow
Specimen collection for this test is optional depending on whether or not the patient has signed an informed consent for them.
1. Specimen Timing:
Bone marrow (BM) will be analysed for minimal residual disease by RT-PCR and immunocytochemistry (ICC) analyses. Marrow specimens for RT-PCR will be obtained at diagnosis, following Cycle 2 induction therapy, end of induction (before transplantation), prior to treatment for MRD and at the end of therapy. ICC analyses will be performed on specimens obtained at the time of diagnosis and at the end of induction (following induction cycle 6) only.
2. Specimen Collection:
Diagnosis and End of Induction:
Specimen Collection: Obtain BM aspirate ( 5 mL ) from each iliac crest and place into separate anticoagulant (Sodium Heparin) tubes. Aspirate and anticoagulant are gently mixed by inverting the tube 3 to 5 times. Withdraw 0.5 mL of one sample (either sample can be used but label side obtained from on shipment form) and place immediately into a PAXgene blood RNA tube (Becton and Dickinson, cat. No. 762165), mix gently.
All Other Time Points: Obtain BM aspirate ( 0.5 mL ) from one iliac crest (either sample can be used but label side obtained from on shipment form) and place immediately into a PAXgene blood RNA tube (Becton and Dickinson, cat. No. 762165), mix gently.
Children's Oncology Group
Label sodium heparin tubes (from applicable time points) with the Biopathology Center (BPC) Number, COG number, specimen type, collection date, Birth Date and include this information on the transmittal forms for immunocytochemical analyses. The sodium heparin tubes should be shipped overnight to Dr.Seeger's lab (see below) at room temperature for immunocytological analysis.
Label the PAXgene tubes with the BPC number, COG patient ID, specimen type and collection date. The PAX gene tube for RT-PCR analysis may be shipped to BPC at room temperature or stored at and below for future shipment. If tubes are to be kept at temperatures below , freeze them first at for 24 hours, then transfer them to or . Ship frozen specimens on dry ice.
3. Shipping Instructions for RT-PCR Specimen
The PAXgene RNA tube can be shipped at room temperature to arrive at the reference laboratory in less than 48 hours. Alternatively, samples can be stored frozen at (see below) and shipped on dry ice as single samples or batched. Ship via Federal Express Priority Overnight using the BPC Federal Express Account number (1290-2562-0). If the specimen is collected on a Friday and will be shipped room temperature, Saturday delivery is available. When a specimen is shipped on Friday, please mark For Saturday Delivery on the air bill and contact the BPC with the tracking number before shipping. Specimens that are frozen should be held and shipped for a weekday delivery.
Institutions are expected to purchase PAXgene tubes for use in ANBL0532 and other COG studies. They are a PreAnalytiX product and can be purchased through their website (catalog #762165; www.preanalytix.com; or through the US vendor VWR, catalog #77776-026; www.vwrsp.com). If PAXgene tubes are not available at your institution and can not be obtained in a timely fashion, please call the Biopathology Center at (800) 347-2486 or BPCBank@Nationwidechildrens.org to arrange delivery for patients anticipated to have high-risk neuroblastoma.
4. Shipping Instructions for Immunocytochemistry Sample
The bilateral_bone marrow aspirate samples collected in sodium heparin tubes sample will be shipped overnight at room temperature for immunocytological analysis. Ship via Federal Express Prior Overnight using the COG Federal Express account number (2504-6481-9). Specimens may be shipped to CHLA on Monday - Thursday for Tuesday - Friday delivery.
Shipping Address:
Robert C. Seeger, M.D.
Neuroblastoma Biology Reference Laboratory
Smith Research Tower, Room #509
Children's Hospital of Los Angeles
4546 Sunset Blvd.
Los Angeles, CA 90027
Telephone: (323) 361-5630
Fax :( 323) 361-3889
Specimen collection for this test is optional depending on whether or not the patient has signed an informed consent for them.
15.5.1 PBSC RT-PCR analysis
Specimen Collection: Withdraw 1 cc PBSC (Day 1 collection) and place into sodium heparin tube. PBSC sample is gently mixed by inverting heparin tube 3 to 5 times
. Withdraw sample from tube and place immediately into 2 PAXgene blood RNA tubes, 0.5 cc per tube.
Label tubes with the BPC Number, COG Patient ID, specimen type and collection date.
Shipping Instructions: RT-PCR Specimen
The PAXgene RNA tube can be shipped at room temperature to arrive at the reference laboratory in less than 48 hours. Alternatively, samples can be stored at and below for future shipment. If tubes are to be kept at temperatures below , freeze them first at for 24 hours, then transfer them to or . Ship frozen specimens on dry ice as single samples or batched. Ship via Federal Express Priority Overnight using the BPC Federal Express Account number (1290-2562-0).
If the specimen is collected on a Friday and will be shipped room temperature, Saturday delivery is available. When shipping a specimen on a Friday, please mark For Saturday Delivery on the air bill and contact the BPC with the tracking number before shipping. Specimens that are frozen should be held and shipped for a weekday delivery.
Specimen collection for this test is optional depending on whether or not the patient has signed an informed consent for them.
Although several studies have shown that the number of circulating neuroblastoma cells is approximately 2 log higher in bone marrow than in peripheral blood, peripheral blood samples should also be screened for MRD because sampling peripheral blood is less invasive and better tolerated by most patients. Specimen will be obtained at diagnosis, end of induction (before transplantation), prior to treatment for MRD (end consolidation) and at the end of therapy.
RT-PCR Analysis:
Peripheral Blood: 2 mL is drawn and placed into an anticoagulant (Sodium heparin or EDTA) tube. Peripheral blood and anticoagulant are gently mixed by inverting the tube 3 to 5 times. The specimen is then withdrawn and placed immediately into a PAXgene blood RNA tube. Label tube with the BPC Number, COG Patient ID, specimen type and collection date. On PAXgene tube also include type of anticoagulant used (Heparin or EDTA).
Shipping: PAXgene blood RNA tubes can be shipped at room temperature to arrive at the reference laboratory in less than 48 hours. Alternatively, samples in PAXgene blood RNA tubes can be stored at and below for future shipment. If tubes are to be kept at temperatures below , freeze them first at for 24 hours, then transfer them to or . Ship frozen specimens on dry ice as single samples or batched. Ship via Federal Express Priority Overnight using the BPC
Federal Express Account number (1290-2562-0). If the specimen is collected on a Friday and will be shipped room temperature, Saturday delivery is available. When shipping a specimen on a Friday, please
mark For Saturday Delivery on the air bill and contact the BPC with the tracking number before shipping. Specimens that are frozen should be held and shipped for a weekday delivery.
Specimen collection for this test is optional depending on whether or not the patient has signed an informed consent for them.
cis-RA Pharmacokinetics and Pharmacogenomics Sample Collection and Shipping
Sample Collection:
ALL SAMPLES MUST BE PROTECTED FROM LIGHT AT ALL TIMES BY WRAPPING IN FOIL IMMEDIATELY.
One blood sample will be collected into sodium heparin tube hours after administration of cisRA on Day 14 of Course 1 of cis-RA treatment. Unseparated blood and plasma must be protected from light at all times (i.e. wrapped in aluminum foil). The blood will be immediately chilled in an ice-water bath and plasma will be separated by low speed centrifugation (1500-2000 rpm for 15 minutes at ). The plasma will be transferred to a polypropylene tube, immediately frozen and stored at wrapped in foil for pharmacokinetic drug analysis. The cell pellet (RBC and WBC) will be frozen and stored at for the pharmacogenomic studies. Each tube must be labeled with patient identifier, study number, date and time the sample was drawn. Data should also be recorded on the 13 Cis Retinoic Acid Pharmacokinetic Shipping (PKS) Form (RDE) to be returned with the samples. Data will also be collected to indicate whether cis-RA was taken by capsule or out of capsule (i.e., capsules snipped and contents mixed with fatty food). Do not label the tubes or PKS form with the patient's name.
Shipment of Samples
Send samples on dry ice WRAPPED IN FOIL, (with a copy of the PKS Form) prepaid via overnight Federal Express, (Account 2504-6481-9), Monday through Thursday WITHIN ONE MONTH OF OBTAINING SAMPLE to:
C Patrick Reynolds, MD PhD
Cancer Center Core Labs STOP 9450
Texas Tech University Health Sciences Center
3601 4th Street
Lubbock, TX 79430-6540
Office Phone: 806-743-1558
Fax: 806-743-2691
Email: PATRICK.REYNOLDS@TTUHSC.EDU
Lab phone: 806-743-2707
Contact in lab: TITO WOODBURN
Email: TITO.WOODBURN@TTUHSC.EDU
(Batch shipping of several patients is encouraged to decrease costs. TTUHSC will collect and batch the samples for PK analysis.
Description of Assay
Plasma concentrations of cis-RA, ATRA and the metabolite 4-oxo-13-cis-RA, will be determined by HPLC analysis. DNA obtained from this sample will be genotyped for metabolising enzymes thought to be involved in the metabolism of cis-RA, such as CYP2C8 and CYP3A7, using PCR methodology
15.8 Cellular Immunity Against Neuroblastoma
Specimen collection for this test is optional depending on whether or not the patient has signed an informed consent for them.
Two samples will be collected for assessments of T-cell responses to neuroblastoma and the tumor antigen Survivin. The first will occur at diagnosis (prior to Cycle 1 of chemotherapy), and the second at the end of consolidation prior to maintenance therapy (after radiation). Ideally, this should occur prior to initiation of Isotretinoin (Accutane) therapy or enrollment on ANBL0032 or ANBL0931.
Sample collection: 20cc peripheral blood in a sodium heparin tube at room temperature. Label the tubes with the COG Patient ID, specimen type and collection date.
Shipment of Samples
Send samples at room temperature. To limit thermal fluctuation, ship in a Styrofoam container with a ROOM TEMPERATURE "ice pack". Do NOT freeze specimen or use a frozen ice pack. Ship overnight for arrival Monday - Friday.
The samples have to be sent with the specimen transmittal form available in the COG website at: https://members.childrensoncologygroup.org/prot/generic.asp.
Ship via overnight Federal Express (2504-6481-9) to:
Stephan Grupp
Division of Oncology
3006 CTRB,
Children's Hospital of Philadelphia
3501 Civic Center Blvd.
Philadelphia, PA 19104
Phone 215-590-5475
Cell 215-847-2957
Description of assays
The central intent of these studies is to assess survivin as a tumor antigen in neuroblastoma. Three immunoassays are available to these studies to measure functional cellular and antitumor immunity . These assays depend on the ability to isolate and utilize autologous antigen presenting cells (APCs) to activate and expand antigen-specific T cells. We have used a system of RNA transfection into engineered antigen presenting cells to examine immune responses in patients with neuroblastoma and to develop a system to engineer cellular cancer vaccines applicable to pediatric tumors . The RNA transfection uses the Amaxa technology, and is capable of transfecting RNA driving the expression of specific proteins such as the tumor antigen survivin as well as tumor RNA to express potential tumor antigens. The APC technology utilizes RNA-loaded CD40-activated B cells (CD40-B) as an alternative APC to induce cytolytic T lymphocyte (CTL) responses. We have shown that we can use these techniques to detect antigen-specific CTL by tetramer staining, demonstrate antigen- or tumor-specific CTL function using ELISPOT or target cell cytolysis, and detect antigen specific CTL using tetramers. Routine assays necessary for these assessments have been established in the lab and the Penn Human Immunology Core (HIC) . These assays include: (i) Flow cytometric T cell assays using monoclonal antibodies and HLA-A2/peptide tetramer complexes, (ii) ELISPOT assay for cytokine release from single peptidespecific T cells, and (iii) T cell cytotoxicity assays in response to survivin expressing targets. This combination of immunoassessment assays will permit the evaluation of whether survivin-specific CD8+
Children's Oncology Group
cells in patients both before after HSCT can be directly enumerated by peptide/MHC tetramers, proliferate and secrete IFN-gamma, and possess phenotypic and functional characteristics of tumor-lytic CD8+ T cells.
We have used HLA-A2/survivin1M2 tetramer to detect survivin-specific CD8+ CTL in HLA-A2+ patients (about half of patients will be A2+). These cells can be detected in the T cell samples collected at diagnosis, and we can use RNA-transfected CD40-B cells as APC to expand anti-tumor CD8+ T cells from patient samples. We will compare the numbers of survivin+ CTL generated using total autologous tumor RNA transfected into CD40-B cells to CTL generated using survivin mRNA. From the initial sample, CD40-B will be generated and electroporated with total autologous tumor RNA or survivin mRNA. After coculture with autologous patient PBMC for one week, tetramer analysis will be performed. Survivin-specific CD8+ CTL will be measured using our HLA-A2/survivin1M2 tetramer, comparing the percentage of CD8+/tetramer+ cells in cultures stimulated with either survivin mRNA or total tumor RNA, with GFP-stimulated cultures serving as negative controls. We will measure anti-tumor CTL cytotoxicty using the standard chromium -release assay. For cytotoxicity assays, HLA-matched allogenic cell lines will be used as targets, with HLA-mismatched neuroblastoma cells as negative control targets. We will also use the CD107a mobilization assay as a marker of CTL function .
16.0 IMAGING STUDIES REQUIRED AND GUIDELINES FOR OBTAINING
Timing of protocol therapy administration, response assessment studies, and surgical interventions are based on schedules derived from the experimental design or on established standards of care. Minor unavoidable departures (up to 72 hours) from protocol directed therapy and/or disease evaluations (and up to 1 week for surgery) for valid clinical, patient and family logistical, or facility, procedure and/or anesthesia scheduling issues are acceptable per COG administrative Policy 5.14 (except where explicitly prohibited within the protocol).
16.1 CT Scans
Axial imaging of the site of the primary tumor will be performed using low-dose technique according to the ALARA concept. The studies will be performed using current-generation single or multi-detector systems. CT slice thickness should be 5 mm or less. Imaging will be performed during the administration of intravenous contrast, generally at . The use of oral contrast will be determined by the individual radiologist performing the study, but may be helpful in abdominal imaging. Images will be reconstructed in soft tissue and edge-enhanced bone/lung and liver algorithms. Coronal and sagittal multiplanar reconstructions may be helpful.
For all patients who achieve CR at end of induction therapy, pre-surgical and end of induction CT scans (or MRI if used instead of CT) will be submitted for central review
16.2 MRI Scans
Typically MRI will be performed on 1.5 T MRI units. Axial and at least one additional plane (coronal or sagittal) of the primary tumor will be performed using at least two pulse sequences (T1, T2, STIR, FLAIR, in/out phase, post contrast). The use of intravenous gadolinium ( ) will be determined by the radiologist performing the study. Slice thickness will be determined by patient size, and region covered, but should be less than 7 mm . The smallest appropriate coil should be used. Measurements should be made using the same axial sequence best showing the tumor in follow-up for comparisons.
Children's Oncology Group
MRI is superior to CT in characterizing epidural tumor extension or leptomeningeal disease, and is the preferred imaging modality in such cases (neck, chest, nonadrenal retroperitoneum) with spinal cord or canal encroachment. It may also be useful in evaluating an MIBG-avid focus detected in the skeleton or soft tissues.
The lack of ionizing radiation also makes MRI attractive for younger children, but this must be balanced with the need for sedation in these patients. With the exceptions noted above, the choice of MRI or CT will be left to the referring pediatric radiologist.
16.3 Skeletal Scintigraphy
Technetium 99 m methylene diphosphonate scans will include the entire skeleton, including multiple overlapping spot images. Scans will be performed using a large field-of-view gamma camera with a highresolution collimator. The typical dose of Tc- 99 m MDP is , with a minimum dose of 74 MBq. In patients with stage 4 disease, if the diagnostic MIBG and bone scans are concordant for all positive sites, then only MIBG scans, but not bone scans, will be required for follow up.
16.4 MIBG Scintigraphy
I-MIBG studies of the entire body will be performed following pretreatment with supersaturated solution of potassium iodide (SSKI) (typical dose is 1 drop years of age one day prior and two days following I-MIBG administration, with a maximum of 3 drops/day). This isotope has significantly less risk to the thyroid gland compared to -MIBG. A typical dose of -MIBG is . Planar and, as appropriate, SPECT imaging will be performed. In patients with stage 4 disease, if the diagnostic MIBG and bone scans are concordant for all positive sites, then only MIBG scans, but not bone scans, will be required for follow up.
16.5 Plain Film Radiography
This will be performed as needed using ALARA technique. If gonadal shielding does not interfere with diagnostic accuracy, it should be utilized. Breast shielding should also be utilized in scoliosis screening unless clinically there is a reason why it should be omitted. In patients with paraspinal disease on MRI at presentation, scoliosis will be evaluated by upright (or supine, if necessary) spine x-ray and measured according to Cobb angle, at presentation and at follow-up (see Section 7.3).
16.6 Tumor Measurement
Tumors will be measured according to the COG radiology group guideline. Diameter of a "measurable mass" must be at least twice the reconstructed slice thickness. Target lesions at baseline must be greater than 1 cm . When multiple or metastatic masses are present, all masses will be described, and up to 5 target masses will be measured using the same method in subsequent follow-ups.
Measurement of each mass (see Appendix V) will consist of the maximal perpendicular diameters ( x and y dimensions) on the slice showing the largest surface area. The length ( z axis) will then be determined by either (a) the difference in table position of the first and last slices showing the tumor plus one slice thickness or (b) the product of [slice thickness + gap] times the number of slices showing the tumor minus one gap distance. According to the elliptical model, the lesion volume is then calculated as 0.5 x [length x width x height].
16.7 Central Review of Imaging
As quality assurance, central review of CT/MRI scans will be performed to correlate with institutional end-induction response assessment. Central review of diagnosis, pre-surgical and end-induction CT and/or MRI studies will be performed for all patients who are considered to have a gross total resection of primary tumor ( residual soft tissue density AND negative MIBG avidity at primary tumor site) OR who are coded by the treating institution as a residual soft tissue density and normal
MIBG scan) at the primary site at completion of induction therapy. CT/MRI studies should be submitted to QARC within 2 weeks of end-induction restaging evaluation. Results of central review will be performed by Day 35 of Consolidation therapy and results will be forwarded to treating institution prior to start of local radiotherapy planning.
Images should preferably be submitted electronically using DICOM format to QARC. Alternatively, they may be submitted on CD using DICOM format. The third option would be hard copy submission of images including soft tissue (abdomen) windows as well as edge-enhanced lung, liver, and bone windows (for CT), or hard copy submission of all pulse sequences obtained (for MRI). Studies should not be photographed smaller than 20 images/page.
16.7.1 QARC address and contact information
Radiology images should be forwarded to:
QARC
272 West Exchange St, Ste 101
Providence, RI 02903-1025
(401) 4544301
17.0 RADIATION THERAPY GUIDELINES
Timing of protocol therapy administration, response assessment studies, and surgical interventions are based on schedules derived from the experimental design or on established standards of care. Minor unavoidable departures (up to 72 hours) from protocol directed therapy and/or disease evaluations (and up to 1 week for surgery) for valid clinical, patient and family logistical, or facility, procedure and/or anesthesia scheduling issues are acceptable per COG administrative Policy 5.14 (except where explicitly prohibited within the protocol).
Radiation Therapy for patients on COG protocols can only be delivered at approved COG RT facilities (see COG Administrative Policy 3.9)
General Guidelines
Radiation therapy will be given following myeloblative stem cell transplantation. Radiation will be given to the primary tumor site and metastatic sites as outlined in this section. The primary site will be irradiated even if complete resection was achieved. Given the high rates of local recurrence following incomplete surgical resection, this study includes boost irradiation of gross residual disease for patients following an incomplete surgical resection. In summary, all patients will receive 21.6 Gy to the primary site. Those with residual disease after induction therapy will receive an additional 14.4 Gy for a total dose of 36.0 Gy .
Required Benchmark and Questionnaires:
Conventional (CT-based 2-dimensional) radiation therapy using photons or electrons, 3D-Conformal Radiotherapy (3D-CRT) and Intensity Modulated Radiation Therapy (IMRT) using photons will be allowed in this study. Patients may not receive intraoperative radiation therapy or proton therapy on this protocol. Centers participating in this protocol using 3D-CRT are required to complete the 3D Benchmark; those using IMRT must complete the IMRT questionnaire and benchmark or phantom (see Section 17.8). Benchmark materials and questionnaires may be obtained from the Quality Assurance Review Center (www.qarc.org) and must be submitted before patients on this protocol can be evaluated. Contact the RPC (http://rpc.mdanderson.org/rpc) for information regarding their IMRT phantoms.
Guidelines and Requirements for the Use of IMRT
Investigators using IMRT will be required to comply with the guidelines developed for the use of IMRT in National Cancer Institute sponsored cooperative group trials. These guidelines are available through www.qarc.org. These guidelines require that the protocol explicitly state their requirements and methods for localization and immobilization; the use of volumetric imaging; target and organ motion management; nomenclature, definitions and rationale for targets and organs at risk; target volume coverage and normal tissue dose constraints; effects of heterogeneity in tissues; and quality assurance.
17.1 Indications for Radiation Therapy
17.1.1 Treatment Sites and Dose
All patients will be irradiated and volumetric targeting for radiation therapy planning should be priority.
Table 17.1.1
Site or Volume (see Section 17.4)
Dose (see Section 17.5)
Primary tumor site (PTV1)
21.6Gy
Residual primary tumor after induction/surgery (PTV2)
14.4Gy
Metastatic disease after induction
21.6Gy
Emergent treatment for organ or life-threatening disease
Contact Study RT Coordinators
17.1.2 Tailoring Fields by Site
Unless constrained in meeting target volume coverage requirements and organ at risk dose recommendations, the dose to the entire vertebral body should be uniform to avoid growth asymmetry. No pelvic irradiation should be given prior to bone marrow or stem cell harvest.
Table 17.1.2
Treatment Site(s)
Methods to anatomically confine CTV
Intra-Abdominal: adrenal and paraspinal ganglia primary sites, involved nodal and metastatic disease
Adjust CTV to avoid kidneys, liver, vertebral bodies
Cervico-Thoracic:
Adjust CTV to avoid uninvolved heart, lung and vertebral bodies
Pelvic:
Adjust CTV to avoid bowel, bladder, and uninvolved bone
Head and Neck:
Adjust CTV to avoid uninvolved critical structures and bone
Footnote: for further details, review special site considerations (Section 17.4.3) and recommendations for organs at risk (Section 17.7).
17.1.3 Timing of Radiation Therapy
Radiation will be given post-stem cell transplant and should start no sooner than 28 days post transplant. Organ toxicity within radiation field should have resolved. Radiotherapy should be initiated within 42 days after stem cell transplantation.
17.1.4 Criteria to Start Radiation Therapy
Table 17.1.4
Organ System
Criteria to Start Radiation Therapy
Hematologic
Absolute phagocyte count
No requirement for platelet count
Mucous Membranes
Mucosa/clinical exam Grade 2 (TPN and enteral feeds are allowed)
Liver Irradiation (Any)
ALT times normal (Grade 2 or less)
Bilirubin times ULN (Grade 2 or less)
No evidence of sinusoidal-obstruction syndrome
Tracheal Irradiation (Any)
Patient stable on room air
< Grade 2 airway edema.
Abdominal Irradiation (Any)
albumin
No albumin infusions for 1 week.
Kidney Irradiation*
serum creatinine should be x normal for age
Renal scintigraphy**
Bladder Irradiation
No hematuria
*Can be achieved with growth factor support.
**Renal scintigraphy is recommended but not required for patients with two functioning kidneys when of one kidney will be irradiated.
17.2 Emergency Irradiation
Patients are allowed to have received radiation therapy at diagnosis to sites of life-threatening or functionthreatening disease. This information must be reported as a part of the quality assurance data. This treatment will not be considered part of the planned primary radiation course that is delivered at a later time provided normal tissue dose constraints are met. Potential deviations from normal tissue dose constraints should be discussed with the study RT coordinators. Emergency irradiation is subject to the same data submission requirements as the primary site (Section 17.8).
17.3 Equipment
Modality: X-rays with a nominal energy of . In the unusual circumstance of a superficial lesion, electron fields may be used. Conventional, conformal, and IMRT techniques are allowed in this study.
Patients may not receive intraoperative radiation therapy or proton beam therapy on this protocol.
Calibration: The calibration of therapy machines used in this protocol shall be verified by the Radiological Physics Center (RPC).
CT treatment planning: All patients will undergo CT treatment planning for this protocol. Slices no more than 5 mm thick is recommended) shall be taken throughout the extent of the irradiated volume.
17.4 Target Volumes
The International Commission on Radiation Units and Measurements (ICRU-50 and 62) prescription methods and nomenclature shall be utilized for this study. This will apply to conformal and nonconformal techniques. Although the post-surgical CT is used for treatment planning, the target volume is based on the extent of disease on pre-operative CT, with modifications noted below, regardless of extent and timing of the surgical resection. If the primary tumor was grossly resected at diagnosis, the radiation therapy should be given to the primary site based on the initial diagnostic tumor volume. No pelvic irradiation should be given prior to bone marrow/PBSC harvest.The GTV, CTV and PTV and normal tissues must be outlined on all CT slices in which the structures exist. Beam's eye view display must be used to design beam apertures.
17.4.1 Primary Site
Gross Tumor Volume 1 (GTV1)
The GTV1 is the volume of tissue containing the highest concentration of residual tumor cells.
The GTV1 includes disease defined by CT, MR and MIBG imaging PRIOR to surgery.
The GTV1 includes disease (tumor and lymph nodes) identified intra-operatively.
The GTV1 is corrected volumetrically after surgical resection but not at the point of attachment.
The GTV1 does NOT include the extent of disease PRIOR to chemotherapy.
The GTV1 does NOT include uninvolved draining lymph node regions.
Special Circumstances (GTV1)
If the primary tumor was grossly resected at diagnosis, GTV1 will be based on the initial diagnostic tumor volume.
In cases where there is discrepancy between imaging studies or intraoperative findings, the larger volume will define GTV1.
When the primary tumor expands into a body cavity such as the lung or displaces a normal structure such as the liver without infiltration, if following surgical resection the normal structure now occupies the space previously occupied by tumor, normal tissue volume should not be included within GTV1.
Gross Tumor Volume 2 (GTV2)
The GTV2 is defined as the volume of residual tumor AFTER induction surgery and chemotherapy measuring
The GTV2 includes disease defined by CT, MR and MIBG imaging.
Special Circumstances (GTV2)
GTV2 will NOT be altered even when there is a complete response after consolidative chemotherapy.
Clinical Target Volume 1 (CTV1)
The CTV is defined as the volume of tissue containing subclinical microscopic disease:
The CTV1 margin should be an expansion of the GTV1 to encompass microscopic disease.
The CTV1 for this protocol is the GTV1 with an anatomically confined margin of 1.5 cm .
The CTV1 should be tailored at tissue interfaces where invasion/infiltration is not likely.
Clinical Target Volume 2 (CTV2)
The CTV is defined as the volume of tissue containing subclinical microscopic disease surrounding the post-surgical residual tumor (GTV2).
The CTV2 margin should be an expansion of the GTV2 to encompass microscopic disease.
The CTV2 for this protocol is the GTV2 with an anatomically confined margin of 1.0 cm .
The CTV2 should be tailored at tissue interfaces where invasion/infiltration is not likely.
Planning Target Volume (PTV1)
The PTV1 is a geometric concept and includes a margin surrounding the CTV1.
The PTV1 should account for physiologic change or motion in the CTV1 and set-up uncertainty.
The PTV1 is defined as the CTV1 with a geometric margin of .
The PTV1 may vary depending on immobilization and cooperation, 0.5 cm is the minimum extent of the margin surrounding CTV1 to form PTV1.
The PTV1 margin does not have to be uniform in all dimensions, especially if it compromises normal tissue volumes or if directional target or normal tissue motion is assessed and understood.
ANBL0532
Planning Target Volume (PTV2)
The PTV2 is a geometric concept and includes a margin surrounding the CTV2.
The PTV2 should account for physiologic change or motion in the CTV2 and set-up uncertainty.
The PTV2 is defined as the CTV2 with a geometric margin of .
The PTV2 may vary depending on immobilization and cooperation, 0.5 cm is the minimum extent of the margin surrounding CTV2 to form PTV2.
The PTV1 margin does not have to be uniform in all dimensions; especially if it compromises normal tissue volumes or if directional target or normal tissue motion is assessed and understood.
Motion Management and Margins to Account for Target Volume and Organ Motion
Considering motion of normal tissues and target volumes is important. The internal target volume (ITV) is defined as the CTV surrounded by the IM component of the PTV and is meant to account for potential motion of the CTV. The planning organ at risk volume (PRV) includes the OR surrounded by a margin to compensate for physiologic change in the target volume. If adequate clinical data do not exist to define the IM component of the PTV or the PRV margin, the following suggestions are provided:
A margin of at least 0.5 cm should be added to any OR to form the PRV.
For a CTV susceptible to physiologic motion, a margin of at least 0.5 cm should be added to the CTV prior to PTV margin expansion or a PTV margin of 1.0 cm should be chosen.
For tumors of the thorax or abdomen, an assessment should be made to determine the extent of motion present. PTV margins should include this motion as a component.
IMRT may be used for tumors of the thorax only if the degree of tumor motion is assessed and can be limited to 0.5 cm in any direction. If required to achieve this goal, techniques for managing or suppressing tumor motion shall be applied.
A description of the method used and evidence (i.e., observed motion during fluoroscopy, motion of surrogate markers using camera systems, or analysis of 4D CT) of the remaining tumor motion should be submitted with the Quality Assurance Documentation materials as noted in section 17.8.
17.4.2 Metastatic Sites
Criteria for Treatment of Metastases: While the primary site is always irradiated, radiation is only given to those metastatic sites with persistent active disease demonstrated on the pre-HSCT evaluation (after Cycle #6 of induction). Sites that are negative on the pre-HSCT scans will NOT be irradiated, even if they had enhanced uptake on MIBG and/or bone scan at diagnosis.
For patients with MIBG positive metastatic sites on pre-HSCT evaluation
If the patient had persistently positive MIBG metastatic sites identified after Cycle #6 of induction, the MIBG scan should be repeated on Day post-HSCT. Only sites still MIBG positive posttransplant should be irradiated. If there are still MIBG positive sites contact Radiation Therapy Study Coordinator to discuss treatment plan prior to starting radiation.
In patients with MIBG positive lesions prior to transplant, consideration should be given to reserving a portion of the collected PBSC product to use as a boost after radiation if counts fall and fail to recover 2+ weeks after radiation is completed. THE STUDY CHAIR OR VICE CHAIR MUST BE NOTIFIED BEFORE REINFUSING PBSC BOOST.
The planning target volume for metastatic sites is the area of residual tumor defined on MIBG, CT, or MR scan with a 1 cm margin. In cases where there is a discrepancy in volume between the scans, the larger volume will be irradiated. For osseous metastases, the margin need not extend more than 1 cm outside the bone or across a joint space.
ANBL0532
17.5 Target Dose
Table 17.5 Prescribed Dose and Fractionation
Nominal Dose by Site
Target Volume
Dose/fraction
Number of Fractions
Primary Site 21.6 Gy
PTV1
1.8 Gy
12
Primary Site Boost 14.4 Gy
PTV2
1.8 Gy
8
Metastatic Sites 21.6 Gy
1.8 Gy
12
Prescribed dose: If conventional methods are used, the prescription point should be at or near the centroid of the target volume. If 3DCRT or IMRT is used, dose should be prescribed to an isodose surface that encompasses the PTV and allows the dose uniformity requirements to be satisfied as noted below.
Dose Definition: Dose is to be specified in centigray (cGy)-to-muscle.
Tissue Heterogeneity: Heterogeneity corrections are required even when conventional planning methods are used and shall be applied for IMRT in compliance with current guidelines for the use of IMRT in clinical trials (guidelines available at www.QARC.org). When IMRT is used in lung, the heterogeneity correction algorithm must be approved by QARC. For questions about heterogeneity corrections or approved algorithms, please contact QARC (www.QARC.org).
Prescription Dose and Fractionation:
Primary Site: The total dose to the PTV will be 21.6 Gy given in 12 fractions. The patient will be treated with one fraction per day, all fields each day, giving 1.8 Gy per fraction to the prescription volume. For patients undergoing an incomplete surgical resection ( residual soft tissue density) as assessed at the end of induction scans, a boost of 14.4 Gy will be delivered to the gross-residual volume for a total dose of 36 Gy.
Metastatic Sites: Sites of persistent active metastatic disease will be irradiated concurrently with the primary site once daily to a total dose of 21.6 Gy given in 12 equal fractions of 1.8 Gy .
Dose Uniformity: For 2D treatment, the dose variations shall be within of the prescription dose. For volume-based treatment plans, the entire PTV should be encompassed within the isodose surface and no more than of the PTV should receive greater than of the prescription dose as evaluated by DVH. Wedges, compensators, and other methods of generating more uniform dose distributions are encouraged.
Interruptions, Delays and Dose Modifications: There will be no planned rests or breaks from treatment, and once radiation therapy has been initiated, treatment will not be interrupted except for any life threatening infection or severe hematologic toxicity defined as or platelets less than during the course of treatment. Under these circumstances, radiation therapy shall be delayed until the counts have recovered. Blood product support should be instituted according to institutional/protocol guidelines. The reason for any interruptions greater than 3 treatment days should be recorded in the patient's treatment chart and submitted with the QA documentation. There should be no modifications in dose fractionation due to age or field size. If any area has been previously treated (emergently), care should be taken not to exceed normal tissue tolerance levels.
17.6 Treatment Technique
Beam Configuration: Every attempt should be made to minimize dose to organs at risk without compromising coverage of the target volume. Although simple opposed field plans are acceptable, the use of oblique fields, or 3-dimensional conformal therapy (coplanar or non-coplanar), or IMRT is encouraged to minimize dose to normal surrounding structures.
Patient Position: Reproducible setups are critical and the use of immobilization devices is strongly encouraged. Use of anesthesia is permitted if necessary for proper positioning.
Field Shaping: Field shaping shall be done with either customized cerrobend blocking or multileaf collimation.
17.7 Organs at Risk
The organ at risk guidelines in this section are recommendations. If the recommended doses to the organs at risk are exceeded because of target volume coverage requirements or other conditions (See Section 17.1.2), an explanation should be included in the quality assurance documentation. In some cases, IMRT may be the preferred treatment method to meet these recommendations and the required target volume coverage guidelines.
Peritoneal Cavity: When the entire peritoneal cavity must be irradiated, the volume to be treated extends from the diaphragmatic domes to the level of the bottom of the obturator foramina. The principles of tailoring fields by site should be considered (Table 17.1.2) and the recommendations for organs at risk should be followed.
Liver: The liver dose should be minimized to reduce the risk of sinusoidal-obstruction syndrome (SOS), formerly known as veno-occlusive disease. No more than of the liver shall receive a cumulative dose greater than 900 cGy and no more than of the liver shall receive a dose greater than 1800 cGy.
Kidney: The kidney doses should be minimized to reduce the risk of chronic renal failure.
If the primary tumor demonstrates laterality to the right or left side, an ipsilateral and contralateral kidney should be designated. In such a case, of the ipsilateral kidney may receive 1440 cGy and of the same kidney may receive 1980 cGy. For the contralateral kidney, no more that should receive a cumulative dose greater than 800 cGy and no more than should receive a cumulative dose greater than 1200 cGy .
If the primary site does NOT demonstrate laterality, both kidneys may receive a mean dose of 14.4 Gy and no more than 50% of each kidney should receive 1980 cGy.
Lung: When a major portion of both lungs must be treated because of a large intrathoracic tumor volume, effort should be made to minimize the irradiated lung volume. No more than one-third of the entire lung volume shall receive a cumulative dose greater than 1500cGy.
17.8 Dose Calculations and Reporting
If 3D conformal techniques are used to treat patients on this study, a 3D benchmark needs to be completed and submitted to QARC. Institutions treating with IMRT must complete the IMRT Questionnaire and either the QARC Benchmark or irradiate the RPC's IMRT head and neck phantom.
ANBL0532
The Benchmark material can be obtained from the Quality Assurance Review Center (www.QARC.org). Contact the RPC (http://rpc.mdanderson.org/rpc) for information regarding their IMRT phantoms. Patients will be considered unevaluable if approved benchmarks are not on file at QARC.
Prescribed Dose:
The monitor units required to deliver the prescribed dose shall be calculated and submitted using the RT-1 or IMRT Dosimetry Summary Form.If IMRT is used, the monitor units generated by the IMRT planning system must be independently checked prior to the patient's first treatment. Measurements in a QA phantom can suffice for a check as long as the patient's plan can be directly applied to a phantom geometry. The total prescribed dose shall be calculated and reported on the RT-2 Radiotherapy Total Dose Record. If 3DCRT or IMRT is used, dose should be prescribed to an isodose surface that encompasses the PTV and allows the dose uniformity requirements to be satisfied.
Dose Uniformity
The maximum and minimum doses in the PTV shall be calculated and reported on the RT-1 or IMRT Dosimetry Summary form. These doses may be extracted from isodose distributions, calculated separately or derived from DVH data.
Normal Tissue Dosimetry
The daily dose to the critical organs indicated should be calculated whenever they are directly included in a radiation field. For 2D techniques they should be reported on the RT-1 Dosimetry Summary form. The total dose shall be calculated and reported on the RT-2 Radiotherapy Total Dose Record form. For patients treated with volume-based techniques, the appropriate dose-volume histograms should be submitted and RT-1 or IMRT form completed. If IMRT is used, a DVH must be submitted for a category of tissue called "unspecified tissue," which is defined as tissue contained within the skin, but which is not otherwise identified by containment within any other structure.
Table 17.8 Required normal tissue DVH data according to primary treatment site
Treatment Area
Required DVH
Neck
Thyroid
Chest
Right Lung
Left Lung
Heart
Abdomen
Liver
Right Kidney
Left Kidney
17.9 Quality Assurance Documentation
On-treatment review is NOT required for this study. Within one week of the completion of radiation therapy, detailed treatment data shall be submitted for the primary site; only the RT-2 form and a copy of the radiotherapy record (treatment chart) need to be submitted for the metastatic site(s).
Please submit the following for the Primary Site Target Volume:
Treatment Planning System Output:
Digitally reconstructed radiographs (DRR) or simulator films for each treatment field and orthogonal (anterior/posterior and lateral) images for isocenter localization for each group of concurrently treated beams. When using IMRT, orthogonal isocenter images are sufficient.
Isodose distributions for the composite treatment plan in the axial, sagittal and coronal planes at the center of the treatment or planning target volume. The planning target volume, isocenter and the normalization method must be clearly indicated.
Dose volume histograms (DVH) for the composite treatment plan for all target volumes and required organs at risk. This shall include GTV1, CTV1, PTV1, and GTV2, CTV2 and PTV2, when indicated. A DVH shall be submitted for the organs at risk specified in section 17.8. When using IMRT, a DVH shall be submitted for a category of tissue called "unspecified tissue." This is defined as tissue contained within the skin, but which is not otherwise identified by containment within any other structure.
Treatment planning system summary report that includes the monitor unit calculations, beam parameters, calculation algorithm, and volume of interest dose statistics.
Beams-eye-view (BEV) of portals showing collimator, beam aperture, target volume and critical structures are required when not using IMRT.
Room-eye-view (REV), if available from the planning system, illustrating all treatment beams and their angles.
Digital Data:
Submission of the treatment plan in digital format is required. Please refer to www.QARC.orghttp://www.QARC.org under "Digital Data" for guidelines regarding digital submission. All submissions, including those that are digital, require hard copy submission of the other items included in this list.
Supportive Data:
All diagnostic imaging used to plan the target volume. This includes CT, MRI and MIBG scans PRIOR to attempted surgical resection of the primary tumor. Digital format is preferred.
Radiotherapy record (treatment chart) including prescription and daily and cumulative doses to all required areas and organs at risk.
Documentation of an independent check of the calculated dose when IMRT is used.
Description of the method used to account for respiratory motion should be documented for review by QARC (when indicated in section 17.4.1).
If the recommended doses to the organs at risk are exceeded, an explanation should be included for review by the QARC and the radiation oncology reviewers.
If emergency RT is administered, documentation should be provided in the form of the RT2 Total Dose Record Form and the radiotherapy record (treatment chart).
Forms:
RT-1 or IMRT Dosimetry Summary Form.
The RT-2 Radiotherapy Total Dose Record Form.
Please submit the following for Metastatic Sites:
Forms:
The RT-2 Radiotherapy Total Dose Record Form.
Radiotherapy record (treatment chart) including prescription and daily and cumulative doses to all required areas and organs at risk.
These data should be forwarded to:
Quality Assurance Review Center
Children's Oncology Group
272 West Exchange St., Suite 101
Providence, Rhode Island 02903-1025
Phone: (401) 454-4301
FAX: (401) 4544683
Questions regarding the dose calculations or documentation should be directed to:
COG Protocol Dosimetrist
Quality Assurance Review Center
272 West Exchange St., Suite 101
Providence, Rhode Island 02903-1025
17.10 Definitions of Deviations in Protocol Performance
Prescription Dose:
Minor Deviation: The prescribed dose differs from that in the protocol by between and
Major Deviation: The prescribed dose differs from that in the protocol by more than
Dose Uniformity:
Minor Deviation for 3D conformal and IMRT treatments:
The entire PTV is not encompassed within the isodose surface representing 95% of the prescription dose or more than of the PTV receives more than of the prescription dose.
Minor Deviation for 2D treatments:
The dose variation in the treated volume shall be within of the prescription point dose.
Volume:
Minor Deviation: Margins less than specified or fields excessively large as deemed by the study reviewer.
Major Deviation: A portion of the tumor (GTV) or potentially tumor bearing area (CTV) is not included in the treated volume.
Critical structures:
A minor or major deviation will be assessed at the time of data review (depending on the details of each case) if the critical structure dose limits are exceeded.
18.0 HEMATOPOIETIC TRANSPLANT GUIDELINES
All transplants performed on COG trials must occur at COG-accredited HSCT programs with the exception of adolescents/adults being treated on COG trials who are referred to an adult transplant facility. See the COG Administrative Policy 3.3 regarding the agreement requirements for these cases.
19.0 RECOMMENDED PROCEDURE FOR PBSC MOBILIZATION AND COLLECTION
19.1 Catheter Use
PBSC may be collected using a large bore double lumen central venous catheter that will allow the flow rates required for apheresis. Many institutions use temporary or tunneled apheresis catheters (such as the 8 Fr Medcomp catheter) in neuroblastoma patients. Femoral line placement is generally not required.
19.2 PBSC Mobilization
Institutional standard operating procedures (SOPs) will be used for mobilization and pheresis. Patients should begin G-CSF starting one day after completing a cycle of chemotherapy. They should continue on G-CSF day while recovering from the cycle of chemotherapy until the post-nadir ANC > 500, at which point it is recommended to increase the dose of G-CSF to at least day per institutional policies.
Institutions which time collections using circulating CD34 cell counts will generally begin when the count is cells . Otherwise, the timing of collection is often within 1-3 days of increasing the GCSF dose, when WBC is (usually Day 14 from start of chemotherapy), although rapid hematopoetic recovery as indicated by a significant left shift and/or a rapidly rising WBC count, may provide an opportunity for earlier collection. It is critical that G-CSF be given daily until PBSC collection is complete. If the WBC is , decrease G-CSF dose to 5 micrograms .
If patients are off G-CSF prior to planned PBSC harvest, they should receive G-CSF , max dose 600 mcg , for 3 days prior to the first day of scheduled PBSC harvest (timing and cytokine regimens per institutional policies), harvest on Day 4 of G-CSF treatment, and continue daily G-CSF until PBSC collections are completed. Such patients should be off their G-CSF for 5-7 days before restarting it for mobilization. If the WBC is , decrease G-CSF dose to 5 micrograms .
19.3 PBSC Collection
19.3.1 Laboratory Studies
For patients , a type and cross for PRBC should be performed one day prior to procedure to avoid apheresis delays.
19.3.2 Apheresis Machine
The Cobe Spectra or the Fenwal CS 3000+ is recommended because the continuous flow centrifugation devices are better tolerated than discontinuous flow machines. Institutional SOPs will be used.
19.3.3 Blood Priming
For patients less than 25 kg , priming of the apheresis machine with IRRADIATED, leukocyte-poor red blood cells may be required.
19.3.4 Collection Goals
It is recommended that large volume apheresis be performed on all patients for each collection. During each leukopheresis procedure, the typical target volume of whole blood processed will be approximately (6 blood volumes).
Optimal collection goal (total for all collections) is CD cells for PBSC. The targeted number of cells can usually be obtained in 1-3 collection days. It is recommended that the collection be stored in at least 3 aliquots of cells:
CD34+ cells/kg for the first HSCT procedure
CD34+ cells/kg for the second HSCT procedure
cells/kg as a backup for delayed engraftment, or for potential subsequent use. Because of the advantages of collecting these backup cells during this pheresis episode (as opposed to later in the patient's clinical course), it is highly recommended that these cells be collected, even if this requires an additional day of apheresis.
CD 34+ cell counts should be done at local institution on all collections as per institutional SOPs.
19.4 PBSC Analyses
The following studies should be performed on each PBSC collection:
Culture for bacterial and fungal contamination,
Nucleated cell count and differential,
CD34+ cell enumeration,
4 PCR analysis for tumor cell content (sent to COG Biopath Center) - see Section 15.5 for details.
19.4.1 PBSC timing:
PBSC mobilization is strongly recommended following cycle 2 REGARDLESS of disease status in the marrow. However, should the patient's medical condition prohibit use of apheresis, it is appropriate to delay PBSC mobilization and harvest after subsequent induction therapy.
PBSC IMMUNOCYTOLOGY IS NOT REQUIRED FOR STUDY PARTICIPATION OR CELL INFUSION. An aliquot of each peripheral stem cell collection may be analyzed for tumor cell content by immunocytochemical analyses per institutional standards. In the rare circumstance that a PBSC collection following Cycle 2 induction is shown to be positive by immunocytology for tumor cell content, patient will require another harvest after Cycle 3 or Cycle 4.
19.5 Cryopreservation of PBSC Products
Each aliquot (as detailed above) should be processed and cryopreserved on the day of collection as per Institutional SOPs. These SOPs include the use of dimethyl sulfoxide final concentration in the cryopreservation medium, use of a monitored, controlled-rate freezer, and storage in liquid nitrogen with appropriate monitoring. The goal is to have 3 separate aliquots cryopreserved in separate bags -1 for each HSCT procedure and 1 for a potential backup.
19.6 PBSC Infusion
The PBSC product should NEVER be irradiated prior to infusion.
19.6.1 Premedication
DMSO may cause a histamine-like reaction when infused into the patient. Therefore premedication with Benadryl and Tylenol is recommended.
19.6.2 Thawing of PBSC
PBSC are thawed in a water bath. Only one bag of PBSC should be thawed at a time - when the infusion of one bag is completed, the next bag should be thawed.
Thawed PBSC should be infused as rapidly as tolerated through a central venous catheter. The unit may be infused by gravity, or the cells may be drawn up into a syringe and pushed by trained personnel. Microaggregate filters and leukodepletion filters MUST NOT be used for infusion of PBSC. If a thawed unit appears clumpy or stringy and these particles cannot be dispersed with gentle kneading, the PBSC product could be infused through a standard 170 micron blood filter.
19.6.3 Possible Symptoms During Infusion
Precipitating Factor
hemolyzed red cells
cellular clumps and debris
cold 10% DMSO
microbial contamination
plasma proteins
Karnofsky and Lansky performance scores are intended to be multiples of 10
ECOG (Zubrod)
Karnofsky
Lansky*
Score
Description
Score
Description
Score
Description
0
Fully active, able to carry on all pre-disease performance without restriction.
100
Normal, no complaints, no evidence of disease
100
Fully active, normal.
90
Able to carry on normal activity, minor signs or symptoms of disease.
90
Minor restrictions in physically strenuous activity.
1
Restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature, e.g., light housework, office work.
80
Normal activity with effort; some signs or symptoms of disease.
80
Active, but tires more quickly
70
Cares for self, unable to carry on normal activity or do active work.
70
Both greater restriction of and less time spent in play activity.
2
Ambulatory and capable of all self-care but unable to carry out any work activities. Up and about more than 50% of waking hours
60
Required occasional assistance but is able to care for most of his/her needs.
60
Up and around, but minimal active play; keeps busy with quieter activities.
50
Requires considerable assistance and frequent medical care.
50
Gets dressed, but lies around much of the day; no active play, able to participate in all quiet play and activities.
3
Capable of only limited selfcare, confined to bed or chair more than 50% of waking hours.
40
Disabled, requires special care and assistance.
40
Mostly in bed; participates in quiet activities.
30
Severely disabled, hospitalization indicated. Death not imminent.
30
In bed; needs assistance even for quiet play.
4
Completely disabled. Cannot carry on any self-care. Totally confined to bed or chair.
20
Very sick, hospitalization indicated. Death not imminent.
20
Often sleeping; play entirely limited to very passive activities.
10
Moribund, fatal processes progressing rapidly.
10
No play; does not get out of bed.
ANBL0532
APPENDIX II: SUPPORTIVE CARE GUIDELINES
These are provided for institutional consideration. The Supportive Care Manual of COG (A Altman editor) can also be referenced for a more detailed discussion of treatment recommendations than can be provided in this appendix. Investigator discretion should be used, and individual considerations made for specific patient situations and institutional practices.
During Induction Therapy
1. Anti-Emetics
Give anti-emetics to control nausea and vomiting as per institutional guidelines. Document all concomitant use of dexamethasone during Cycle 1 and 2 on the topotecan pharmacokinetics data collection form as dexamethasone may alter drug metabolism.
2. Cytokine Support
Only for induction cycles 1 and 3-6: Patients will receive Myeloid growth factor (cytokine) support per institutional guidelines beginning 24-48 hours after chemotherapy until ANC post nadir. For eg: Filgrastim (G-CSF) day is given as a single daily Sub Qor IV dose.
Growth factor support should be stopped at least 24 hr before the next chemotherapy cycle. ANC must be and platelets before starting next course of therapy. The ANC frequently falls after discontinuing myeloid growth factor support (secondary neutrophil nadir). If the ANC recovers to after nadir but then falls to after the myeloid growth factor is stopped, the next cycle of therapy can be given despite if all other criteria for the next cycle of chemotherapy are met.
For induction cycle 2 (or subsequent PBSC collection cycle)
During cycle when PBSC are to be harvested, filgrasatim (G-CSF) must be used for myeloid growth factor support. G-CSF should be continued until harvest is completed. See guidelines for G-CSF dosing during PBSC harvest (See Section 19.2).
3. Blood Product Support
Recommend that patients receive prophylactic platelet transfusions as per institutional guidelines. Use irradiated, CMV appropriate products. Patients should have platelet count of on the day of apheresis. May receive a platelet transfusion to reach 50,000 requirement.
Recommend that patients receive packed red blood cell transfusions to maintain their hemoglobin > 7.0 gm/dl. Use irradiated, CMV appropriate products.
Investigators in Canadian institutions need to follow the CSA standards for Blood and Blood Components CAN/CSA-Z902-04 issued in March 2004 and available at http://www.shopcsa.ca.
4. Pneumocystis Prophylaxis
Patients should be given prophylactic co-trimoxazole (sulfamethoxazole/trimethoprim) based on 5 mg trimethoprim/kg/day divided BID orally administered on 3 consecutive days a week. For sulfa-intolerant patients it is recommended that inhaled pentamidine or oral dapsone be used as prophylaxis. Co-
trimoxazole should be held if patients fail to recover hematopoiesis by Day 21 of any cycle. It can be re-started once the next chemotherapy cycle is given or patient can be changed to pentamidine or dapsone if there is concern about continued myelosuppression from co-trimoxazole.
5. Treatment of Fever and Neutropenia
Patients should be admitted to the hospital and treated with antibiotic therapy that is appropriate to that institution's infections experience and antibiotic sensitivity patterns.
6. Herpes Simplex Virus Prophylaxis
Antiviral prophylaxis against HSV is recommended for all patients who are HSV 1 or HSV 2 seropositive. Recommend use of acyclovir day PO divided BID, rounded to the nearest 100 mg (max dose 800 mg PO BID) OR day IV divided q 12 hours (if unable to take PO meds for reasons other than mucositis) to begin on the day of completion of chemotherapy administration, continue throughout neutropenia, and stop when neutropenia (post-nadir ) and mucositis have resolved.
7. Nephrotoxic Agents
Due to risk for nephrotoxicity following dose intensive platinum therapy, minimize use of additional nephrotoxic antibiotic therapy. Whenever possible monitor blood levels of the antibiotics used. When empiric or directed anti-fungal therapy is required, utilize lipid-based amphotericin or non-amphotericin antifungal therapy as clinically indicated for patient.
Supportive Care During BMT
1. Isolation
Protective isolation per local institutional guidelines.
2. Skin Care
Thiotepa can cause significant skin toxicity with sloughing of skin. To avoid skin toxicity, patient should be bathed 4-6 times daily during the three days of Thiotepa administration and the day following. Avoid large occlusive dressings and use of any skin creams and remove adhesive residue from prior dressings and leads. Change sheets with bathing and bathe with water only, avoiding soap. Consider placement of a Foley catheter during the days of Thiotepa use in younger children to avoid perineal burns. Perineal burns may be more related to sweating induced by diapers than contact with urine.
3. Blood Product Support
Patients should receive prophylactic platelet transfusions and packed red blood cell transfusions as per institutional guidelines. Blood products should be irradiated following the current FDA guidelines found at: http://www.fda.gov/cber/gdlns/gamma.htm
Investigators in Canadian institutions need to follow the CSA standards for Blood and Blood Components CAN/CSA-Z902-04 issued in March 2004 and available at http://www.shopcsa.ca.
4. Bacterial Prophlyaxis
Patient will receive empiric broad spectrum antibiotic therapy for treatment of neutropenia or febrile neutropenia per institutional guidelines.
5. Fungal Prophylaxis
Recommend use per institutional guidelines. Fluconazole day daily until neutrophil engraftment is a standard approach.
6. Pneumocystis Pneumonia Prophylaxis
Day -10 to day -2 before BMT: trimethoprim ( ) and sulfamethoxazole ( ); divide into two doses and give q 12 hr . for prophylaxis against P . carinii. After engraftment, administer PCP prophylaxis) per institutional guidelines.
7. Viral Prophylaxis
Prophylaxis against HSV and/or VZV will be given in seropositive patients as per institutional guidelines. In addition, patients should be monitored for EBV and CMV reactivation per institutional guidelines. Recommendation to screen EBV by PCR and CMV as per institutional practice (i.e. antigenemia or PCR) q2-4 weeks through day 120 for CMV+ and/or EBV+patients.
Suggested supportive care during cis-RA therapy
Topical Vitamin E should be applied to lips BID during cis-RA therapy if cheilitis develops. All patients should avoid direct sun exposure while on cis-RA. All patients should avoid exposure to Vitamin A products during cis-RA therapy.
APPENDIX III: NB WITH INTRASPINAL EXTENSION +/- SPINAL CORD COMPRESSION Evaluation and grading of neurologic and orthopedic dysfunction
Neurological symptoms:
Yes No
Duration of neurologic symptoms: days days
L
28 days
Motor deficit*:
Grade 0
None
Grade 1
Mild weakness
Grade 2
Moderate weakness
Grade 3
Severe weakness
Sensory deficit:
Yes No
Sphincter dysfunction^:
Bladder
Yes No
Bowel
Yes No
Pain
Yes No
Orthopedic findings:
Scoliosis (by spine x-ray): None
Mild (<20 degree curve)
Moderate (20-40 degree curve)
Severe (>40 degree curve)
Extremity abnormality:
Leg length discrepancy
Yes No
Foot size discrepancy/foot deformity
Yes No
*Motor Deficit:
Grade I = mild weakness
Legs walking disability
Arms = difficulty raising arms above head
Grade II = moderate weakness
Legs = inability to walk &/or make movements against gravity
Arms = inability to raise arms above head
Grade III severe weakness
Paraplegia
No elicitable DTRs
No muscular movements
Sphincter Deficit:
Distended bladder
Patulous anus
Constant urine dripping
Stress incontinence
Any significant change/deterioration from prior bowel or bladder function
APPENDIX IV: INTERNATIONAL NEUROBLASTOMA STAGING SYSTEM (INSS)
Stage 1
Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically (nodes attached to and removed with the primary tumor may be positive).
Stage 2A
Localized tumor with incomplete gross resection; representative ipsilateral non-adherent lymph nodes negative for tumor microscopically.
Stage 2B
Localized tumor with or without complete gross excision, with ipsilateral non-adherent lymph nodes positive for tumor; enlarged contralateral lymph nodes must be negative microscopically.
Stage 3
Unresectable unilateral tumor infiltrating across the midline , with or without regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement; or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement.
Stage 4
Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, and/or other organs (except as defined for Stage 4S).
Stage 4S
Localized primary tumor (as defined for Stage 1, 2A or 2B) with dissemination limited to skin, liver, and/or bone marrow (limited to infants year of age).
Multifocal primary tumors (e.g., bilateral adrenal primary tumors) should be staged according to the greatest extent of disease, as defined above, and followed by a subscript "M" (e.g. ).
The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column.
Marrow involvement in Stage 4 S should be minimal, i.e., less than of total nucleated cells identified as malignant on bone marrow biopsy or marrow aspirate. More extensive marrow involvement would be considered to be Stage 4. The MIBG scan (if performed) should be negative in the marrow.
Proven malignant effusion within the thoracic cavity if it is bilateral or the abdominal cavity upstages the patient to INSS 3
APPENDIX V: TUMOR SIZE MEASUREMENTS BY CROSS-SECTIONAL IMAGING
RELATIONSHIP BETWEEN CHANGE IN SINGLE DIAMETER (RECIST), PRODUCT OF TWO DIAMETERS (WHO), AND THREE PERPENDICULAR DIAMETERS ("VOLUME")
COG GUIDELINE: TUMOR SIZE MEASUREMENT BASED ON CROSSSECTIONAL IMAGING
, & E are contiguous parallel slices in the X-Y plane (usually axial) showing the tumor
W and T are the maximal perpendicular diameters on the slice ( C in this example) showing the largest surface area
Tumor length in the Z -axis ( L ) (perpendicular to X-Y plane) can be obtained either by the [a] (difference in table position of the first and last slices showing the tumor plus one slice thickness), or [b] the product of ([slice thickness + gap] and the number of slices showing the tumor) minus one gap distance
WHO criteria: TxW is used
RECIST: the larger of the two ( ) is used (W in this example)
Elliptical model volume
The same modality and measurement method used in the initial imaging should be used in follow ups
Target lesions at baseline must measure greater than 1 cm ; if these target lesions decrease in size to below 1 cm , care should be taken in measuring and inadvertently progressing a patient due to minimal changes in measurement from a nadir value below 1 cm , which may be within measurement error. When multiple primary or metastatic masses are present, all masses will be described. However, up to 5 target masses should be measured, using the same method in subsequent follow ups.
APPENDIX VI: NEUROBLASTOMA RESPONSE CRITERIA
(International Recommendations)
SITE
TEST
COMPLETE RESPONSE
VERY GOOD PARTIAL RESPONSE
PARTIAL RESPONSE
MIXED RESPONSE
NO RESPONSE
PROGRESSI VE DISEASE
Primary
3 dimensional CT or MRI imaging (determine volume from product of three dimensions physical exam and/or surgical measurement)
no tumor
>90% reduction in 3-dimensional tumor volume
50-90% reduction in 3dimensional tumor volume
50-90% reduction of any measurable lesion (primary or metastases); no new lesions; <25% increase in any existing lesions, exclude bone marrow evaluation
no new lesions; <25% increase in any lesion; exclude bone marrow evaluation
any new lesion; increase of any measurable lesion by >25%; previous negative bone marrow positive
Metastases
Bone marrow (aspirate x 2 and biopsy x 2)
Bone x-rays and scintigraphy (Tc and/or MIBG)
no lesions
all lesions improved; no new lesions
50-90% reduction
50-90% reduction
Tumor marker
Urine catecholamine metabolites (HVA & VMA)
Normal
normal or both decreased >90%
both decreased 50-90%
Response must be evaluated before and after surgery for the primary site. If complete response, very good partial response, or partial response is achieved surgically, indicate such when reporting the response.
The total response can be no better than the worst response in any subcategory (e.g., if primary complete response, metastases partial response, and, tumor marker very good partial response, the total response partial response).
Immunocytology results are not used to determine response.
One sample may be positive only if there is a reduction in the number of sites originally positive for tumor at diagnosis.
bone scan may show residual abnormalities but the MIBG scan (if performed) must be negative.
bone scan and/or MIBG scan (if performed) must show improvement, but residual abnormalities may be present on either scan.
Measure palpable lymph nodes in 3 dimensions and calculate tumor volume.
Brodeur G et al: JCO 1993; 11:1466-1477
APPENDIX VII: NEUROBLASTOMA STUDY ASSIGNMENT TABLE
Study
Stage
Age
MYCN
Ploidy
Histology
Other
ANBL00B1
1
any
any
any
any
ANBL00B1
2a/2b
any
not amp
any
any
resection
ANBL0531
2a/2b
any
not amp
any
any
resection <50%
ANBL0531
2a/2b
any
not amp
any
any
biopsy only
ANBL0532
2a/2b
any
amp
any
any
any degree of resection
ANBL0531
3
<547d
not amp
any
any
ANBL0531
3
not amp
any
FH
ANBL0532
3
any
amp
any
any
ANBL0532
3
not amp
any
UH
ANBL0532
4
<365d
amp
any
any
ANBL0531
4
<365d
not amp
any
any
ANBL0532
4
365-<547d
amp
any
any
ANBL0532
4
365-<547d
any
any
ANBL0532
4
365-<547d
any
any
UH
ANBL0531
4
not amp
DI
FH
ANBL0532
4
any
any
any
ANBL00B1
4s
<365d
not amp
DI
FH
asymptomatic
ANBL0531
4s
<365d
not amp
any
asymp or symp
ANBL0531
4s
<365d
missing
missing
missing
too sick for biopsy
ANBL0531
4s
<365d
not amp
any
any
symptomatic
ANBL0531
4s
<365d
not amp
any
UH
asymp or symp
ANBL0532
4s
<365d
amp
any
any
asymp or symp
FOOTNOTE:
DI - DNA Index
FH - Favourable (Shimada) Histology
UH - Unfavourable (Shimada) Histology
APPPENIX VIII: TOPOTECAN PHARMACOKINETICS DATA COLLECTION FORM
This form comes with your specimen package form and must be completed and returned with specimens when shipped. ALL FIELDS ARE REQUIRED.
Please note that the PK topotecan evaluation form completed on eRDES is different from this form and ALSO has to be completed.
COG No.:
Acc. No.:
Institution:
Gender:
Race:
Height:
Weight:
BSA:
Topotecan dosage ( ):
Date Administered:
Date of Birth:
Has the patient received prior chemotherapy? Circle one: Yes or No
If yes, please indicate names and dates of all prior chemotherapy on an additional sheet.
2. List the name, dose and regimen of other drugs the patient has received within 48 hours of topotecan therapy (for more space use an additional sheet):
NOTE: YOU ARE REQUIRED TO DOCUMENT ALL CONCOMITANT USE OF DEXAMETHASONE DURING CYCLE 1 AND 2 AS IT MAY ALTER DRUG METABOLISM
Drug Name
Drug Dose
When Administered
Please fill in course and day of therapy, time dose was administered, time blood was scheduled to be drawn and actual time blood was drawn:
Course Day
Scheduled Time
Actual Time
START of infusion
END of infusion
15 minutes after end of infusion ( )
Name of person completing form
Phone:
Date:
Please send this completed form with the sample to:
Stewart Laboratory
Pharmaceutical Sciences Department
Chili's Care Center I5500
St. Jude Children’s Research Hospital
262 Danny Thomas Place
Memphis, TN 38105
Ph: (901) 595-2400
APPENDIX IX: 13-CIS RA PHARMACOKINETIC DATA COLLECTION FORM
13-CIS RA PHARMACOKINETIC DATA COLLECTION FORM
ANBL0532 - Phase III Randomized Trial of Single vs. Tandem Myeloablative Consolidation Therapy for High-Risk Neuroblastoma
DATE OF SUBMISSION:
(Mm/dd/yyyy)
(Mm/dd/yyyy)
COG REG. NO:
Height
Weight:
BSA:
1. Name of the Institution:
2. 13 - cis-RA Administration:
a. Date(mm/dd/yyyy) :
b. Time :
c. Cycle (number):
d. Day of cycle:
3. PK collection:
a. Date(mm/dd/yyyy):
b. Time of collection:
Please check the relevant boxes as directed for Qs 4 and 5:
4 Type of cis-RA administration:
a. Capsule (directly)
b. Out of capsule (i.e., capsules snipped and contents mixed with fatty food) □
If out of capsule, please specify:
Route: □ by mouth, □ NG tube, □ G-Tube, □ other, Fatty foods mixed with drug:
5. Gender
Male: □
Female: □
6. Filled out by:
Name: Contact number:
Signature:
INSTRUCTIONS FOR PK COLLECTION AND SHIPPING:
ALL SAMPLES MUST BE PROTECTED FROM LIGHT AT ALL TIMES BY WRAPPING IN FOIL IMMEDIATELY.
The blood sample ( ) will be collected into sodium heparin tubes hours after administration of cis-RA on Day 14 of Course 1 of cis-RA treatment. Refer to Section 15.7 of the protocol for more details.
Please send this completed form with the sample to:
C Patrick Reynolds, MD PhD
Cancer Center Core Labs STOP 9450
Texas Tech University Health Sciences Center
3601 4th Street, Lubbock, TX 79430-6540
Office Phone: 806-743-1558
Email: Patrick.Reynolds@TTUHSC.edu
Lab phone: 806-743-2707
Contact in lab: TITO WOODBURN
Email: TITO.WOODBURN@TTUHSC.EDU
APPENDIX X: SPECIMEN SUBMISSION SCHEDULE
Required specimens are found in shaded boxes only; all other specimens are optional
Immunocytochemistry
Sections 15.4
Optional (BM at CHLA)
Cellular Immunity (CHOP)
Section 15.8
Optional
RT-PCR (BPC) Sections 15.4 & 15.6
** Contact BPC for questions regarding
PAX Gene tubes Optional
Place in sodium heparin tube (green), mix and transfer to PAX tubes
Contact St.Jude (Stewart Laboratory ) for Topotecan PK kit. If treatment starts before arrival of kit, access Memos on COG website (12-14-07b; 5-8-08b) for instructions on sample acquisition or contact Stewart lab at 901-595-2400. Peripheral blood 2 mL sodium heparin tube (green) 15 +/- 10 minutes after end of topotecan infusion. Send to: St Jude/ Stewart Lab.
Peripheral blood 2 mL sodium heparin tube (green) minutes after end of topotecan infusion. Send to: St Jude/ Stewart Lab.
Induction Cycle 2, END and Day 1 of stem cell collection
1. Bone marrow 0.5 ml in PAX tube
2.1 ml of PBSC product in 2 PAX tubes ( each)
Surgery ( Look)
Required for ANBL00B1: Tumor Tissue to BPC. 1. 2 H&E slides from paraffin block; 2. Representative paraffin blocks or 10 unstained slides from most representative blocks on coated slides for IHC use. 3. Snap frozen tissue 4. Viable fresh tissue
Induction, End
Bone marrow each side in sodium heparin tubes
1. Bone marrow 0.5 ml PAX tube
2. Peripheral blood 2 ml in PAX tube
Consolidation, End
Bone marrow 5 ml each side in sodium heparin tube (green)
ONLY IF ENROLLING onto ANBL0032 or ANBL0931
Peripheral Blood 20ml in sodium heparin tube 60-90D post HSCT
Peripheral Blood 5 ml in sodium heparin tube wrapped in foil 4 hours after cis-RA. Send to: C. Patrick Reynolds, Cancer Center Core Labs STOP 9450, Texas Tech University Health Sciences Center, 3601 4th Street, Lubbock, TX 79430-6540 Office Phone: 806-743-1558 Lab phone: 806-743-2707 Email: Patrick.Reynolds@TTUHSC.edu.
Contact in lab: TITO WOODBURN Email: TITO.WOODBURN@TTUHSC.EDU
End of therapy
1. Bone marrow in PAX tube
2. Peripheral blood 2ml in PAX tube
Relapse
Same specimens as at the time of surgery to be sent to BPC
BPC: COG Biopathology Center, Nationwide Children's Hospital 700 Children's Dr, Rm WA 1340 Columbus OH 43205 _ Ph.no: 614-722-2810.
St Jude: Stewart Laboratory. Chili's Care Center I5500, St Jude Children's Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105.
CHLA: Robert Seeger, Childrens Hospital Los Angeles 4546 Sunset Blvd, NBL Biology Reference Lab, Smith Research Tower-Rm #509,Los Angeles, CA 90027 Ph.no: 323-361-5630.
CHOP: Stephan Grupp Division of Oncology Abramson 902, Children's Hospital of Philadelphia 3615 Civic Center Blvd. Philadelphia, PA 19104 Ph.no: 215-590-.5475
SCCA: Linda Risler, Shen/McCune Lab, 4225 Roosevelt Way NE Suite 100, Seattle WA 98105. Phone prior to shipping: 206-685-1650 Fax: 206-543-3835
Refer to Protocols ANBL00B1 & ANBL0532 for specific collection, processing & shipping instructions
APPENDIX XI: YOUTH SUMMARIES FOR CHILDREN AND TEENS
INFORMATION SHEET REGARDING RESEARCH STUDY ANBL0532
(for children from 7 through 12 years of age)
A Study of Neuroblastoma (that has spread from where it started) Treatment Comparing One versus TwoHigh Dose Chemotherapy Regimens in Consolidation
We have been talking with you about neuroblastoma. Neuroblastoma is a type of cancer that grows mostly in the nervous system outside the brain. We have done special tests, scans and blood tests on you to find out if you have high-risk neuroblastoma. The tests tell us that you have neuroblastoma that is hard to treat because it has spread from where it started.
We are asking you to take part in a research study because you have the type of neuroblastoma that is hard to treat. A research study is when doctors work together to try out new ways to help people who are sick. In this study, we are trying to learn more about how to treat high-risk neuroblastoma. We will do this by:
a. adding a new anti-cancer medicine, called Topotecan, to the usual chemotherapy that children with neuroblastoma receive
b. giving higher doses of radiation (or x-ray treatments) to children who still have tumor after getting induction chemotherapy medicine
c. giving some children one cycle of high dose chemotherapy and some children two cycles of high dose chemotherapy, followed by either one or two stem cell transplants, to see if this will keep the neuroblastoma from coming back after treatment is stopped
d. studying how your body responds to the medicines.
We want to see if these things can help more children can get rid of their neuroblastoma. We don't know if these approaches will help or not. That is why we are doing this study.
3. Children who are part of this study will be treated first with chemotherapy that will include the newer medicine, Topotecan, as well as several other chemotherapy medicines. Chemotherapy is a type of medicine that destroys cancer cells. Some children may also have surgery or radiation to get rid of as much of the neuroblastoma as possible. After this first part of therapy, some children will get one cycle, while others may get two cycles of high dose chemotherapy followed by a stem cell transplant. All children in this study will get the medicine, Accutane, after the chemotherapy, stem cell transplant, surgery and radiation are done. Sometimes you will have extra blood drawn for special research tests.
4. Sometimes good things can happen to people when they are in a research study. These good things are called "benefits." A benefit of being part of this study may be a chance that your cancer will go away for as long as possible. We don't know for sure if there is any benefit of being part of this study. We expect that the information learned from this study will help other patients with high-risk neuroblastoma in the future.
5. Sometimes bad things can happen to people when they are in a research study. These bad things are called "risks." The risks to you from this study are possibly having more side effects or the therapy may not work to make the cancer go away. With this treatment you can have problems, both during and after the treatment. We will check you closely so we can treat the problems you may have. Other things may happen to you that we don't yet know about.
6. Your family can choose to be part of this study or not. Your family can also decide to stop being in this study at any time once you start. There may be other treatments for your illness that your doctor can tell you about. Make sure to ask your doctors any questions that you have.
7. We are asking your permission to collect additional blood and bone marrow specimens. We want to see if there are ways to tell how the cancer will respond to treatment. These samples would be taken when other standard blood tests are being performed, so there would be no extra procedures. This part of the study is optional. You can still take part in this study even if you don't allow us to collect the extra blood or bone marrow samples for research.
INFORMATION SHEET REGARDING RESEARCH STUDY (for teens from 13 through 17 years of age)
A Study of Neuroblastoma (that has spread from where it started) Treatment Comparing One versus Two High Dose Chemotherapy Regimens in Consolidation
We have been talking with you about neuroblastoma. Neuroblastoma is a type of cancer that grows mostly in the nervous system outside the brain. It can also be found anywhere in the body. We have done special tests, scans and blood tests on you to find out if you have high-risk neuroblastoma. The tests tell us that you have neuroblastoma that is hard to treat because it has spread from where it started.
We are asking you to take part in a research study because you have the type of neuroblastoma that is hard to treat. A research study is when doctors work together to try out new ways to help people who are sick. In this study, we are trying to learn more about how to treat neuroblastoma. We will do this by:
a. adding a new anti-cancer medicine, called Topotecan, to the usual chemotherapy that children with neuroblastoma receive
b. giving higher doses of radiation (or x-ray treatments) to children who still have tumor after getting induction chemotherapy medicine
c. giving some children one cycle of high dose chemotherapy and some children two cycles of high dose chemotherapy, followed by either one or two stem cell transplants, to see if this will keep the neuroblastoma from coming back after treatment is stopped in more children
d. studying how your body responds to medicines.
We want to see if these things can help more children can get rid of their neuroblastoma. We don't know if these approaches will help or not. That is why we are doing this study.
3. Children who are part of this study will be treated with chemotherapy, surgery, radiation treatment and stem cell transplant(s). The treatment is given in three phases: Induction, Consolidation and Maintenance. Chemotherapy is a type of strong medicine that destroys cancer cells. The chemotherapy in this study is very strong and may make you feel pretty sick at times. The doctors will give you medicines to help you when you are sick. You will be in the hospital a lot to get the chemotherapy. In the induction phase, you will receive chemotherapy, including the new combination, cyclophosphamide and topotecan and you will also have a procedure(s) called a stem cell harvest. You will be connected to a machine that will remove blood from you, take out the baby cells (stem cells) that grow up to be normal blood cells, and then give the rest of the blood back to you. During consolidation, you will get either one or two cycles of very high doses of chemotherapy, followed by either one or two stem cell transplants. The last phase of therapy is called maintenance. During this phase you will be taking pills by mouth twice a day for two weeks. Then you will have 2 weeks that you do not take the pills. This phase lasts for six months. Throughout the treatment you will have some extra blood draws for special research tests.
4. Sometimes good things can happen to people when they are in a research study. These good things are called "benefits." A benefit of being part of this study may be a chance that your cancer will go away for as long as possible. We don't know for sure if there is any benefit of being part of this study. We expect that the information learned from this study will help other patients with high-risk neuroblastoma in the future.
5. Sometimes bad things can happen to people when they are in a research study. These bad things are called "risks." With this treatment you can have problems, both during and after the treatment. The risks to you from this study involve very low blood counts, possible infections and organ damage. You will be given medicines to help your blood counts recover after the treatment, to prevent infection, and to reduce organ damage. There is also a chance of the cancer cells growing back. We will check you closely so we can treat the problems you may have. Other things may happen to you that we don't yet know about.
6. Your family can choose to be part of this study or not. Your family can also decide to stop being in this study at any time once you start. There may be other treatments for your illness that your doctor can tell you about. Make sure to ask your doctors any questions that you have.
7. We are asking your permission to collect additional blood and bone marrow specimens. We want to see if there are ways to tell how the cancer will respond to treatment. These samples would be taken when other standard blood tests are being performed, so there would be no extra procedures. This part of the study is optional. You can still take part in this study even if you don't allow us to collect the extra blood or bone marrow samples for research.
Children's Oncology Group
REFERENCES
1 Young JR, Miller RW: Incidence of malignant tumors in U.S. children. J Pediatr Surg 86:254-8, 1975
2 Garaventa A, DeBernardi B, Pianca C, et al: Localized but unresectable neuroblastoma: Treatment and outcome of 145 cases. J Clin Oncol 11:1770-9, 1993
3 Kushner BH, O'Reilly RJ, Mandell LR, et al: Myeloablative combination chemotherapy without total body irradiation for neuroblastoma. Journal of Clinical Oncology 9:274-9, 1991
Pinkerton CR, et al: Megatherapy for soft tissue sarcomas: EBMT experience. Bone Marrow Transplant 7:120, 1991
Stram DO, Matthay KK, O'Leary M, et al: Consolidation chemotherapy and autologous bone marrow transplantation vs. continued chemotherapy for metastatic neuroblastoma: A report of two concurrent Children's Cancer Group studies. J Clin Oncol 14:2417-2426, 1996
Frappaz D, Michon J, Coze C, et al: LMCE3 treatment strategy: results in 99 consecutively diagnosed stage 4 neuroblastomas in children older than 1 year at diagnosis. J Clin Oncol 18:46876, 2000
7 Matthay KK, Castleberry RP: Treatment of advanced neuroblastoma: the US experience, (ed 1), Elsevier Science B.V.:417-436, 2000
8 Haas-Kogan DA, Swift PS, Selch M, et al: Impact of radiotherapy for high-risk neuroblastoma: a Children's Cancer Group study. International Journal of Radiation Oncology, Biology, Physics 56:28-39, 2003
9 Schmidt ML, Lukens JN, Seeger RC, et al: Biologic factors determine prognosis in infants with stage IV neuroblastoma: A prospective Children's Cancer Group study. J Clin Oncol 18:1260-8, 2000
Schmidt ML, Lal A, Seeger RC, et al: Favorable prognosis for patients 12 to 18 months of age with stage 4 nonamplified MYCN neuroblastoma: a Children's Cancer Group Study. J Clin Oncol 23:6474-80, 2005
George RE, London WB, Cohn SL, et al: Hyperdiploidy plus nonamplified MYCN confers a favorable prognosis in children 12 to 18 months old with disseminated neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 23:6466-73, 2005
Park J, Villablanca J, Seeger R, et al: Favorable outcome of high risk (HR) stage 3 neuroblastoma (NB) with myeloablative therapy and 13-cis-retinoic acid. Proc ASCO Abstract # 8503, 2005
Matthay KK, Villablanca JG, Seeger RC, et al: Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13 -cisretinoic acid. Children's Cancer Group. New England Journal of Medicine 341:1165-73, 1999
Kushner BH, LaQuaglia MP, Bonilla MA, et al: Highly effective induction therapy for stage 4 neuroblastoma in children over 1 year of age. J Clin Oncol 12:2607-2613, 1994
August CS, Serota FT, Kock PA, et al: Treatment of advanced neuroblastoma with supralethal chemotherapy, radiation, and allogeneic or autologous marrow reconstitution. J Clin Oncol 2:60914, 1984
Flandin I, Hartmann O, Michon J, et al: Impact of TBI on late effects in children treated by megatherapy for Stage IV neuroblastoma. A study of the French Society of Pediatric oncology. Int J Radiat Oncol Biol Phys 64:1424-31, 2006
Ladenstein R, Lasset C, Hartmann O, et al: Comparison of auto versus allografting as consolidation of primary treatments in advanced neuroblastoma over one year of age at diagnosis: report from the European Group for Bone Marrow Transplantation. Bone Marrow Transplantation 14:37-46, 1994
Smedler AC, Bolme P: Neuropsychological deficits in very young bone marrow transplant recipients. Acta Paediatr 84:429-33, 1995
Olshan JS, Willi SM, Gruccio D, et al: Growth hormone function and treatment following bone marrow transplant for neuroblastoma. Bone Marrow Transplantation 12:381-5, 1993
Kolb H J, Bender-Gotze C: Late complications after allogeneic bone marrow transplantation for leukaemia. Bone Marrow Transplant 6:61-72, 1990
McCowage GB, Vowels MR, Shaw PJ, et al: Autologous bone marrow transplantation for advanced neuroblastoma using teniposide, doxorubicin, melphalan, cisplatin, and total-body irradiation. J Clin Oncol 13:2789-95, 1995
Garaventa A, Rondelli R, Lanino E, et al: Myeloablative therapy and bone marrow rescue in advanced neuroblastoma. Report from the Italian Bone Marrow Transplant Registry. Bone Marrow Transplantation 18:125-130, 1996
Ladenstein R, Philip T, Lasset C, et al: Multivariate analysis of risk factors in stage 4 neuroblastoma patients over the age of one year treated with megatherapy and stem-cell transplantation: a report from the European Bone Marrow Transplantation Solid Tumor Registry. J Clin Oncol 16:953-65, 1998
Philip T, Ladenstein R, Zucker JM, et al: Double megatherapy and autologous bone marrow transplantation for advanced neuroblastoma: the LMCE2 study. Brit J Cancer 67:119-127, 1993
Grupp SA, Stern JW, Bunin N, et al: Rapid-sequence tandem transplant for children with highrisk neuroblastoma. Medical & Pediatric Oncology 35:696-700, 2000
Grupp SA, Stern JW, Bunin N, et al: Tandem high-dose therapy in rapid sequence for children with high-risk neuroblastoma. Journal of Clinical Oncology 18:2567-75, 2000
George RE, Li S, Medeiros-Nancarrow C, et al: High-risk neuroblastoma treated with tandem autologous peripheral-blood stem cell-supported transplantation: long-term survival update. J Clin Oncol 24:2891-6, 2006
Buckner CD, Rudolph Rh, Fefer A, et al: High dose cyclophosphamide therapy for malignant disease. Toxicity, tumor response and the effect on stored marrow. . Cancer 29:357-65, 1972
Kletzel M, Abella E, Sandler E, et al: Thiotepa and Cyclophosphamide with Stem Cell Rescue for Consolidation Therapy for Children with High-Risk Neuroblastoma: A Phase I/II Study of the Pediatric Blood and Marrow Transplant Consortium. J Ped Hem Onc 20:49-54, 1998
Herzig RH, Fay JW, Herzig GP, et al: Phase I - II with high-dose thiotepa and autologous marrow transplantation in patients with refractory malignancies. Advances in Cancer Chemotherapy, Park Row Publishers:17-23, 1987
Clamon GH: Alkylating Agents. . 1992
Kretschmar CS, Kletzel M, Murray K, et al: Response to paclitaxel, topotecan, and topotecancyclophosphamide in children with untreated disseminated neuroblastoma treated in an upfront phase II investigational window: a pediatric oncology group study. Journal of Clinical Oncology 22:4119-26, 2004
Pinkerton C, Zucker J, Hartmann O, et al: Short duration, high dose, alternating chemotherapy in metastatic neuroblastoma (ENSG 3C induction regimen). British Journal of Cancer 62:319-323, 1990
Castleberry RP, Cantor AB, Green AA, et al: Phase II investigational window using carboplatin, iproplatin, ifosfamide, and epirubicin in children with untreated disseminated neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 12:1616-20, 1994
Valteau-Couanet D, Michon J, Boneu A, et al: Results of induction chemotherapy in children older than 1 year with a stage 4 neuroblastoma treated with the NB 97 French Society of Pediatric Oncology (SFOP) protocol. J Clin Oncol 23:532-40, 2005
Potmesil M: Camptothecins: from bench research to hospital wards. Cancer Res 54:1431-9, 1994
Houghton PJ, Cheshire PJ, Myers L, et al: Evaluation of 9-dimethylaminomethyl-10-hydroxy camptothecin (topotecan) against xenografts derived from adult and childhood tumors. Ann Oncol 3:84, 1992
Zamboni WC, Stewart CF, Thompson J, et al: Relationship between topotecan systemic exposure and tumor response in human neuroblastoma xenografts. J Natl Cancer Inst 90:505-11, 1998
Kaufmann SH, Peereboom D, Buckwalter CA, et al: Cytotoxic effects of topotecan combined with various anticancer agents in human cancer cell lines. [see comments.]. Journal of the National Cancer Institute 88:734-41, 1996
Chou TC, et al: Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to climical protocal design. JNCI 86:1517, 1994
Zamboni W, Heideman R, Meyer W, et al: Pharmacokinetics of topotecan in pediatric patients with normal and altered renal function. Proceedings of ASCO 15:180, 1996
Tubergen D, Stewart C, CB P, et al: Phase I trial and pharmacokinetic (PK) and pharmacodynamic (PD) study of topotecan using a five-day course in children with refractory solid tumors: A Pediatric Oncology Group study. J Ped Hemat Oncol 18:352-361, 1996
Mattern MR, Hofmann GA, Polsky RM, et al: In vitro and in vivo effects of clinically important camptothecin analogues on multidrug-resistant cells. Oncol Res 5:467-74, 1993
Keshelava N, Groshen S, Reynolds CP: Cross-resistance of topoisomerase I and II inhibitors in neuroblastoma cell lines. Cancer Chemotherapy & Pharmacology 45:1-8, 2000
Stewart CF, Baker SD, Heideman RL, et al: Clinical pharmacodynamics of continuous infusion topotecan in children: systemic exposure predicts hematologic toxicity. JCO 12:1946-1954, 1994
Kramer K, Kushner B, Heller G, et al: Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer 91:1510-9, 2001
Rowinsky EK, Adjei A, Donehower RC, et al: Phase I and pharmacodynamic study of the topoisomerase I-inhibitor topotecan in patients with refractory acute leukemia. J Clin Oncol 12:2193-2203, 1994
Saltz L, Sirott M, Young C, et al: Phase I clinical and pharmacology study of topotecan given daily for 5 consecutive days to patients with advanced solid tumors, with attempt at dose intensification using recombinant graanulocyte colony-stimulating factor. J Natl Canc Inst 85:1499-1507, 1993
Pratt C, Stewart C, Santana V, et al: Phase I study of topotecan for pediatric patients with malignant solid tumors. JCO 12:539-543, 1994
Nitschke R, Parkhurst J, Sullivan J, et al: Topotecan in pediatric patients with recurrent and progressive solid tumors: A Pediatric Oncology Group phase II study. J Ped Hem Onc 20:315318, 1998
Saylors RL r, Stewart CF, Zamboni WC, et al: Phase I study of topotecan in combination with cyclophosphamide in pediatric patients with malignant solid tumors: a Pediatric Oncology Group Study. Journal of Clinical Oncology 16:945-52, 1998
Saylors RL r, Stine KC, Sullivan J, et al: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. Journal of Clinical Oncology 19:3463-9, 2001
Kushner BH, Kramer K, Meyers PA, et al: Pilot study of topotecan and high-dose cyclophosphamide for resistant pediatric solid tumors. Med Pediatr Oncol 35:468-474, 2000
Donato ML, Gershenson DM, Wharton JT, et al: High-dose topotecan, melphalan, and cyclophosphamide (TMC) with stem cell support: a new regimen for the treatment of advanced ovarian cancer. Gynecol Oncol 82:420-6, 2001
Park J, Stewart C, London W, et al: Targeted Topotecan during Induction Therapy of High Risk Neuroblastoma : A COG Pilot Study. Proc ASCO Abstract 9013. 2006
Weiss R: Ifosfamide vs cyclophosphamide in cancer therapy. Oncology 5:67-76, discussion -82, 4-6 1991
Halperin EC: Long-term results of therapy for stage C neuroblastoma. J Surg Oncol 63:172-8, 1996
Rosen EM, Cassady JR, Frantz CN, et al: Neuroblastoma: the Joint Center for Radiation Therapy/Dana-Farber Cancer Institute/Children's Hospital experience. J Clin Oncol 2:719-32, 1984
Matthay KK, Atkinson JB, Stram DO, et al: Patterns of relapse after autologous purged bone marrow transplantation for neuroblastoma: a Childrens Cancer Group pilot study. J Clin Oncol 11:2226-33, 1993
Wolden S, Gollamudi S, Kushner B, et al: Local control with multimodality therapy for stage 4 neuroblastoma. Int J Rad Oncol 46:969-974, 2000
Kushner BH, Wolden S, LaQuaglia MP, et al: Hyperfractionated low-dose radiotherapy for highrisk neuroblastoma after intensive chemotherapy and surgery. Journal of Clinical Oncology 19:2821-8, 2001
Bradfield SM, Douglas JG, Hawkins DS, et al: Fractionated low-dose radiotherapy after myeloablative stem cell transplantation for local control in patients with high-risk neuroblastoma. Cancer 100:1268-75, 2004
West DC, Shamberger RC, Macklis RM, et al: Stage III neuroblastoma over 1 year of age at diagnosis: improved survival with intensive multimodality therapy including multiple alkylating agents. J Clin Oncol 11:84-90, 1993
Castleberry RP, Kun LE, Shuster JJ, et al: Radiotherapy improves the outlook for patients older than 1 year with Pediatric Oncology Group stage C neuroblastoma. J Clin Oncol 9:789-95, 1991
Jacobson HM, Marcus RB J, Thar TL, et al: Pediatric neuroblastoma: postoperative radiation therapy using less than 2000 rad. Int J Radiat Oncol Biol Phys 9:501-5, 1983
Simon T, Bongartz R, Hero B, et al: Intensified External Beam Radiation Therapy Improves the Outcome of Stage 4 Neuroblastoma in Children Year with Residual Local Disease. Advances in Neuroblastoma Research:Abstract 314, 2006
Adkins ES, Sawin R, Gerbing RB, et al: Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg 39:931-6, 2004
de Bernardi B, Rogers D, Carli M, et al: Localized neuroblastoma. Surgical and pathologic staging. Cancer 60:1066-72, 1987
De Bernardi B, Balwierz W, Bejent J, et al: Epidural compression in neuroblastoma: Diagnostic and therapeutic aspects. Cancer Lett 228:283-99, 2005
Plantaz D, Rubie H, Michon J, et al: The treatment of neuroblastoma with intraspinal extension with chemotherapy followed by surgical removal of residual disease. A prospective study of 42 patients--results of the NBL 90 Study of the French Society of Pediatric Oncology. Cancer 78:311-9, 1996
Hayes FA, Green AA, O'Connor DM: Chemotherapeutic management of epidural neuroblastoma. Med Pediatr Oncol 17:6-8, 1989
Katzenstein HM, Kent PM, London WB, et al: Treatment and outcome of 83 children with intraspinal neuroblastoma: the Pediatric Oncology Group experience. . J Clin Oncol 19:1047-55, 2001
De Bernardi B, Pianca C, Pistamiglio P, et al: Neuroblastoma with symptomatic spinal cord compression at diagnosis: treatment and results with 76 cases. J Clin Oncol 19:183-90, 2001
Reynolds CP, Kane DJ, Einhorn PA, et al: Response of neuroblastoma to retinoic acid in vitro and in vivo. Prog Clin Biol Res 366:203-211, 1991
Reynolds CP, Schindler PF, Jones DM, et al: Comparison of 13-cis-retinoic acid to trans-retinoic acid using human neuroblastoma cell lines. Progress in Clinical & Biological Research 385:23744, 1994
Sidell N, Altman A, Haussler MR, et al: Effects of retinoic acid (RA) on the growth and phenotypic expression of several human neuroblastoma cell lines. Experimental Cell Research 148:21-30, 1983
Thiele CJ, Reynolds CP, Israel MA: Decreased expression of N-myc precedes retinoic acidinduced morphological differentiation of human neuroblastoma. Nature 313:404-6, 1985
Olson JA: Adverse effects of large doses of vitamin A and retinoids. Seminars in Oncology 10:290-3, 1983
Villablanca JG, Khan AA, Avramis VI, et al: Hypercalcemia: a dose-limiting toxicity associated with 13-cis-retinoic acid. American Journal of Pediatric Hematology-Oncology 15:410-5, 1993
McCune JS, Slattery JT: Pharmacological considerations of primary alkylators. Cancer Treat Res 112:323-45, 2002
Tasso MJ, Boddy AV, Price L, et al: Pharmacokinetics and metabolism of cyclophosphamide in paediatric patients. Cancer Chemother Pharmacol 30:207-11, 1992
Estlin EJ, Veal GJ: Clinical and cellular pharmacology in relation to solid tumours of childhood. Cancer Treat Rev 29:253-73, 2003
Yule SM, Price L, McMahon AD, et al: Cyclophosphamide metabolism in children with nonHodgkin's lymphoma. Clin Cancer Res 10:455-60, 2004
Yule SM, Boddy AV, Cole M, et al: Cyclophosphamide pharmacokinetics in children. British Journal of Clinical Pharmacology 41:13-19, 1996
Yule SM, Price L, Cole M, et al: Cyclophosphamide metabolism in children following a and a 24-h infusion. Cancer Chemotherapy & Pharmacology 47:222-8, 2001
Ren S, Kalhorn TF, McDonald GB, et al: Pharmacokinetics of cyclophosphamide and its metabolites in bone marrow transplantation patients. Clin Pharmacol Ther 64:289-301, 1998
Fasola G, Lo Greco P, Calori E, et al: Pharmacokinetics of high-dose cyclophosphamide for bone marrow transplantation. Haematologica 76:120-5, 1991
Moore MJ, Hardy RW, Thiessen JJ, et al: Rapid development of enhanced clearance after highdose cyclophosphamide. Clin Pharmacol Ther 44:622-8, 1988
Graham MI, Shaw IC, Souhami RL, et al: Decreased plasma half-life of cyclophosphamide during repeated high-dose administration. Cancer Chemother Pharmacol 10:192-3, 1983
Ren S, Yang JS, Kalhorn TF, et al: Oxidation of cyclophosphamide to 4hydroxycyclophosphamide and deschloroethylcyclophosphamide in human liver microsomes. Cancer Res 57:4229-35, 1997
Lamba V, Lamba J, Yasuda K, et al: Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (constitutive androstane receptor) expression. J Pharmacol Exp Ther 307:906-22, 2003
Rettie A: Cyp2c, in Levy R, Thummel K, Trager W, et al (eds): Metabolic Drug Interactions. Philadelphia, Lippincott Willams & Williams.75-86, 2000
Sullivan-Klose TH, Ghanayem BI, Bell DA, et al: The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 6:341-9, 1996
Sweeney C, Ambrosone CB, Joseph L, et al: Association between a glutathione S-transferase A1 promoter polymorphism and survival after breast cancer treatment. Int J Cancer 103:810-4, 2003
Chang TK, Yu L, Maurel P, et al: Enhanced cyclophosphamide and ifosfamide activation in primary human hepatocyte cultures: response to cytochrome P-450 inducers and autoinduction by oxazaphosphorines. Cancer Res 57:1946-54, 1997
Petros W, Hopkins P, Vredenburgh J, et al: Associations Between Variants in Several Drug Metabolism Genes and Chemotherapy Pharmacokinetics or Clinical Response. Proc AACR 42:1435, 2001
Villablanca JG, Khan AA, Avramis VI, et al: Phase I trial of 13-cis-retinoic acid in children with neuroblastoma following bone marrow transplantation. J Clin Oncol 13:894-901, 1995
Khan AA, Villablanca JG, Reynolds CP, et al: Pharmacokinetic studies of 13-cis-retinoic acid in pediatric patients with neuroblastoma following bone marrow transplantation. Cancer Chemother Pharmacol 39:34-41, 1996
Veal G, Errington J, Koller K, et al: Pharmacokinetics and metabolism of 13-cis-retinoic acid in children with neuroblastoma. Proc. AACR 44:3554 (abstract), 2003
Marrill J, Capron CC, Idres N, et al: Human cytochrome P450s involved in the metabolism of 9-cis- and 13-cis-retinoic acids. Biochemical Pharmacology 63:933-943, 2002
McSorley LC, Daly AK: Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem Pharmacol 60:517-26, 2000
Dai D, Zeldin DC, Blaisdell JA, et al: Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 11:597-607, 2001
Bahadur N, Leathart JB, Mutch E, et al: CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem Pharmacol 64:1579-89, 2002
Samokyszyn VM, Gall WE, Zawada G, et al: 4-hydroxyretinoic acid, a novel substrate for human liver microsomal UDP-glucuronosyltransferase(s) and recombinant UGT2B7. J Biol Chem 275:6908-14, 2000
Bhasker CR, McKinnon W, Stone A, et al: Genetic polymorphism of UDPglucuronosyltransferase 2B7 (UGT2B7) at amino acid 268: ethnic diversity of alleles and potential clinical significance. Pharmacogenetics 10:679-85, 2000
Santana VM, Furman WL, Billups CA, et al: Improved response in high-risk neuroblastoma with protracted topotecan administration using a pharmacokinetically guided dosing approach. J Clin Oncol 23:4039-47, 2005
Stewart CF, Iacono LC, Chintagumpala M, et al: Results of a phase II upfront window of pharmacokinetically guided topotecan in high-risk medulloblastoma and supratentorial primitive neuroectodermal tumor. J Clin Oncol 22:3357-65, 2004
Andersen MH, Thor SP: Survivin--a universal tumor antigen. Histol Histopathol 17:669-75, 2002 Islam A, Kageyama H, Takada N, et al: High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 19:617-23, 2000
Coughlin CM, Fleming MD, Carroll RG, et al: Immunosurveillance and survivin-specific T cell immunity in children with high-risk neuroblastoma, in press. . J. Clin. Oncol, 2006
cytolytic T cells. Nat Med 9:1377-82, 2003
Powell JL, Bunin NJ, Callahan C, et al: An unexpectedly high incidence of Epstein-Barr virus lymphoproliferative disease after CD34+ selected autologous peripheral blood stem cell transplant in neuroblastoma. Bone Marrow Transplant 33:651-7, 2004
Cheung NK, Von Hoff DD, Strandjord SE, et al: Detection of neuroblastoma cells in bone marrow using GD2 specific monoclonal antibodies. J Clin Oncol 4:363-9, 1986
Favrot MC, Frappaz D, Maritaz O, et al: Histological, cytological and immunological analyses are complementary for the detection of neuroblastoma cells in bone marrow. Br J Cancer 54:63741, 1986
Gussetis ES, Ebener U, Wehner S, et al: Immunological detection and definition of minimal residual neuroblastoma disease in bone marrow samples obtained during or after therapy. Eur J Cancer Clin Oncol 25:1745-53, 1989
Combaret V, Favrot MC, Kremens B, et al: Immunological detection of neuroblastoma cells in bone marrow harvested for autologous transplantation. Br J Cancer 59:844-7, 1989
Faulkner LB, Tintori V, Tamburini A, et al: High-sensitivity immunocytologic analysis of neuroblastoma cells in paired blood and marrow samples. J Hematother 7:361-6, 1998
Moss TJ, Reynolds CP, Sather HN, et al: Prognostic value of immunocytologic detection of bone marrow metastases in neuroblastoma. N Engl J Med 324:219-26, 1991
marrow RC Reynold CP Galleg R et al Q11
Seeger RC, Reynolds CP, Gallego R, et al: Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group Study. J Clin Oncol 18:4067-76, 2000
Burchill SA, Selby PJ: Molecular detection of low-level disease in patients with cancer. J Pathol 190:6-14, 2000
Reynolds CP, Moss TJ, Seeger RC, et al: Sensitive detection of neuroblastoma cells in bone marrow for monitoring the efficacy of marrow purging procedures. Prog Clin Biol Res 175:42541, 1985
Piantadosi S: Clinical Trials: A Methodologic Perspective. Hoboken, NJ, John Wiley and Sons, Inc.:687, 2005
Brodeur GM, Pritchard J, Berthold F, et al: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11:1466-77, 1993
Shimada H, Ambros IM, Dehner LP, et al: Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. . Cancer 86:349-63, 1999
Shimada H, Ambros IM, Dehner LP, et al: The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 86:364-72, 1999
Cheung IY, Sahota A, Cheung NK: Measuring circulating neuroblastoma cells by quantitative reverse transcriptase-polymerase chain reaction analysis. . Cancer 101:2303-8, 2004
Coughlin CM, Vance BA, Grupp SA, et al: RNA-transfected CD40-activated B cells induce functional T-cell responses against viral and tumor antigen targets: implications for pediatric immunotherapy. Blood 103:2046-54, 2004
Coughlin CM, Vonderheide RH: Targeting adult and pediatric cancers via cell-based vaccines and the prospect of activated B lymphocytes as a novel modality. Cancer Biol Ther 2:466-70, 2003
Boone JM, Geraghty EM, Seibert JA, et al: Dose reduction in pediatric CT: a rational approach. Radiology 228:352-60, 2003
Kushner BH: Neuroblastoma: a disease requiring a multitude of imaging studies. J Nucl Med 45:1172-88, 2004
Siegel MJ, Ishwaran H, Fletcher BD, et al: Staging of neuroblastoma at imaging: report of the radiology diagnostic oncology group. Radiology 223:168-75, 2002
Shapiro B, Gross MD: Radiochemistry, biochemistry, and kinetics of 131Imetaiodobenzylguanidine (MIBG) and 123I-MIBG: clinical implications of the use of 123IMIBG. Med Pediatr Oncol 15:170-7, 1987
Grupp SA, Cohn SL, Wall D, et al: Hematopoietic Stem Cell Transplant Discipline and the Neuroblastoma Disease Committee, Children's Oncology Group.Collection, storage, and infusion of stem cells in children with high-risk neuroblastoma: saving for a rainy day. Pediatr Blood Cancer 46(7):719-22, 2006
SAMPLE INFORMED CONSENT / PARENTAL PERMISSION FOR PARTICIPATION IN RESEARCH
This model informed consent form has been reviewed by the DCT/NCI and is the official consent document for this study. Local IRB changes to this document are allowed. (Institutions should attempt to use sections of this document which are in bold type in their entirety.) Editorial changes to these sections may be made as long as they do not change information or intent. If the local IRB insists on making deletions or more substantive modifications to the risks or alternatives sections, they must be justified in writing by the investigator and approved by the IRB .
ANBL0532, Phase III Randomized Trial of Single vs. Tandem Myeloablative Consolidation Therapy for High-Risk Neuroblastoma
PART 1: CONSENT FOR INDUCTION THERAPY
When the word "you" appears in this consent form, it refers to you or your son or daughter; "we" means the doctors and other staff.
WHY ARE YOU BEING INVITED TO TAKE PART IN THIS STUDY?
This is a clinical trial, a type of research study. Your study doctor will explain the clinical trial to you.
The Children's Oncology Group (COG) is carrying out this study. COG is an international research group that conducts clinical trials for children with cancer at more than 200 hospitals in the United States, Canada, Australia, New Zealand, the Netherlands, and Switzerland. It is common medical practice to treat children with cancer on research studies like this one.
You are being asked to take part in this study because you have high risk neuroblastoma. Neuroblastoma is a type of cancer. Neuroblastoma shows up as a lump or mass in the belly or around the spinal cord in the chest, neck, or pelvis. Neuroblastoma is a cancer of nerve cells. It develops in nerve cells that are outside of the brain. It often spreads to bone, liver, lymph nodes and bone marrow, which is the soft tissue in the center of bones where blood cells are made. You have the type of neuroblastoma that is called High Risk because your tumor has spread from where it started or because your type of tumor is harder to treat.
It is common to enroll children and adolescents with cancer in a clinical trial that seeks to improve cancer treatment over time. Clinical trials include only people who choose to take part. You have a choice between a standard treatment for High Risk neuroblastoma and this clinical trial. Please take your time to make your decision about taking part in this clinical trial. You may discuss your decision with your friends and family. You can also discuss it with your health care team. We encourage you to include your child in the discussion and decision to the extent that she or he is able to understand and take part. If you have any questions, you can ask your study doctor for more explanation.
WHAT IS THE CURRENT STANDARD OF TREATMENT FOR THIS DISEASE?
The treatment for neuroblastoma includes 3 parts (phases) of therapy called Induction, Consolidation and Maintenance. During Induction therapy anti-cancer drugs (chemotherapy) and surgery are used to kill and remove as much tumor as possible. Blood stem cells are collected during the Induction phase of therapy. After collection the blood stem cells are frozen and stored to be used during Consolidation phase of treatment. Blood stem cells are the cells that create new blood cells, such as red blood cells, white blood cells, and platelets. Blood stem cells can be collected from the blood by using a machine that can separate out the part of blood that has stem cells and then return the remaining blood back to the patient.
During the Consolidation phase of treatment extremely high doses of chemotherapy are given to better kill any remaining neuroblastoma cells. The extremely high doses of chemotherapy destroy healthy bone marrow. Bone marrow is the soft tissue in the hollow of flat bones of the body that produces new blood cells. The peripheral blood stem cells that were stored during the Induction phase of treatment are given back to the patient after the high dose Consolidation chemotherapy. These stems cells allow the bone marrow to return to normal so that new blood cells can be made. This type of therapy is called a Hematopoietic Stem Cell Transplant. Once the patient has healed from the effects of high doses of chemotherapy, radiation therapy is given to the first place the tumor was found and to any additional places where the tumor was found after Induction therapy.
During the third phase of therapy, Maintenance, an oral drug, called cis-retinoic acid (Accutane) is given.
WHY IS THIS STUDY BEING DONE?
This study is being done because more than half of the patients with high risk neuroblastoma will not be cured of their disease.
The main purpose of this research study is to find out if using 2 cycles of high dose Consolidation chemotherapy, instead of 1 cycle, will decrease the chance that the neuroblastoma will grow back after therapy. This research study will also find out if adding a new anti-cancer drug to Induction therapy will decrease the number of patients who have tumor present at the end of the Induction phase of therapy. This research study will also find out if patients whose tumor does not go away with initial therapy will need a higher dose of radiation therapy.
The treatment will be described in two separate consent forms. Part 1 will discuss the use of a drug added to the standard anti-cancer drugs used in the Induction phase of neuroblastoma treatment. It will also explain standard blood stem cell collection and surgery. Part 1 of the consent is being given to you now.
Your doctor will give you Part 2 (the second consent form) of the study to you after you have completed the Induction phase of therapy. You will not be able to participate in Part 2 of this study if your tumor has grown larger or spread to new areas in your body. The second consent (Part 2) will discuss the Consolidation and Maintenance phases of therapy. You do not have to take part in Part 2 of the study. You can discuss this with your doctor and make that decision after you have finished the Induction therapy.
Part 1 of this study will collect information about your neuroblastoma and about the effects of the first phase of treatment, Induction which lasts about 20 weeks ( 5 months).
Part 1 of this study will test the addition of the combination of chemotherapy, cyclophosphamide/topotecan, to the standard chemotherapy combinations used in the Induction phase of treatment for high risk neuroblastoma. The use of cyclophosphamide and topotecan during induction phase of treatment is experimental. This study is designed to help understand whether this new treatment will increase our ability to kill or remove your tumor cells.
During this part of the study, we will:
Look at how well high risk neuroblastoma responds to this new Induction therapy.
Study how certain genes (genes direct activities of cells) affect how you respond to the cyclophosphamide and topotecan induction chemotherapy.
Study whether your immune system (blood cells that help fight infection) can produce cells that will find and kill neuroblastoma.
Study how topotecan is broken down by your body.
Study whether new tests can be used to find small amounts of neuroblastoma cells.
HOW MANY PEOPLE WILL TAKE PART IN THIS STUDY?
About 664 subjects will take part in this study. The study will last about years.
WHAT WILL HAPPEN ON THIS STUDY?
You will need to have the following exams, tests or procedures to find out if you can be in the study and to check how you are doing during treatment. These exams, tests or procedures are part of regular cancer care and may be done even if you do not join the study. If you have had some of them recently, they may not need to be repeated. This will be up to your study doctor.
Physical exam
Blood tests
Bone marrow tests
Pregnancy test for females of childbearing potential prior to treatment
Various scans (x-ray tests to determine whether your tumor is responding to therapy)
Tests of kidney function
Tests of lung and heart function
Hearing tests
Urine Tests
A diagram of treatment can be seen below.
Diagram of treatment
Induction Chemotherapy
This treatment uses chemotherapy to make the tumor as small as possible, hopefully allowing it to be removed. All patients will begin treatment with the Induction phase of chemotherapy, which is divided into six 3 -week cycles. There are three different drug combinations used during the six cycles. Cycles 1 and 2 will use the new combination of cyclophosphamide and topotecan. Cycles 3 and 5 use the anti-cancer drugs etoposide and cisplatin. Cycles 4 and 6 use the anti-cancer drugs vincristine, cyclophosphamide, and doxorubicin and the drugs MESNA. MESNA is given to help protect the bladder from potential damaging effects of cyclophosphamide. The drugs used Cycles 3, 4, 5 and 6 are standard drugs used for the treatment of high risk neuroblastoma study. Cycles of chemotherapy will be given approximately every 21 days. An additional drug referred as the myeloid growth factor (eg: filgrastim (G-CSF)) is given after the chemotherapy. It helps start the production of white blood cells (infection fighting cells) and will help your body recover from treatment. Any myeloid growth factor may be given after chemotherapy for induction cycles 1 and 3-6 whereas only filgrastim (G-CSF) (granulocyte colony stimulating factor) has to be specifically given after cycle 2 .
Methods for Giving Drugs
Your doctor will recommend the placement of a tube, or catheter, into a large vein in the chest or neck during a short operation as the standard way to give chemotherapy into a vein. This tube is called a central line. You will be given medication to help you sleep during this procedure and pain medication afterwards to help you stay comfortable. The central line is used to give chemotherapy drugs and to take small amounts of blood for testing during treatment. A central line will be used to give chemotherapy even if you do not participate in this study. The risks associated with central lines will be explained to you by your surgeon. You will be given a separate informed consent form to read and sign prior to having a central line inserted.
You may also receive one of your medications as an injection under the skin.
Schedule and Methods of Giving Chemotherapy
A schedule of how and when the drugs will be given during the Induction phase of therapy is listed below.
Various methods will be used to give drugs:.
PO - Drug is given by tablet or liquid swallowed through the mouth (PO).
IV - Drug is given using a needle or tubing inserted into a vein. It can be given by IV push over several minutes or by infusion over minutes or hours
SUBQ - Drug is given by injecting a needle into the tissue just under the skin (SUBQ shot)
Most drugs on this study will be given using a needle or tubing inserted into a vein (IV).
CALENDAR FOR THERAPY
INDUCTION (Total of 6 cycles; one cycle given every 3 weeks ( 21 days)
Drug
Method
Schedule
Cycle 1
Cyclophosphamide
IV infusion for 30 minutes
X
X
X
X
X
Topotecan
IV infusion for 30 minutes
X
X
X
X
X
Myeloid growth factor
Depends on the agent used
X
Day
1
2
3
4
5
6-21
Cycle 2
Cyclophosphamide
IV infusion for 30 minutes
X
X
X
X
X
Topotecan
IV infusion for 30 minutes
X
X
X
X
X
Filgrastim (G-CSF)
Sub Q (or) IV
X
Cycles 3 and 5:
Day
1
2
3
4
5
6-21
Cisplatin
IV infusion for 1 hour
X
X
X
X
Etoposide
IV infusion for 1 hour
X
X
X
Myeloid growth factor
Depends on the agent used
X
Cycles 4, 6:
Day
1
2
3
4
5
6-21
Cyclophosphamide
IV infusion for 6 hours
X
X
MESNA
IV infusion prior to each cyclophosphamide dose and at 4 and 8 hours after each cyclophosphamide dose.
X
X
Doxorubicin
IV infusion for 24 hours
X
X
X
Vincristine
IV infusion for 1-2 minutes
X
X
X
Myeloid growth factor
Depends on the agent used
X
Stem Cell Harvest
After the second cycle of Induction is finished, your stem cells will be collected using a procedure called apheresis. During apheresis, your blood is collected into a machine that filters out the stem cells and the filtered blood is returned to your body. This procedure may need to be done several times to collect enough stem cells for the Consolidation phase of therapy. This procedure will be performed at [local institution]. The stem cells will be frozen and stored until needed for the Consolidation phase of therapy.
The risks of Stem Cell Harvests can be found in Attachment #2.
Surgery
After the fifth cycle of Induction, you will have surgery to remove as much remaining tumor as possible. The surgeon will talk to you about your surgery and any possible risks involved with the surgery.
Additional Medicines
You may get some extra medications, like antibiotics, to help fight infection. Other medications may be given to lessen the side effects of chemotherapy. These medicines are part of standard care.
Additional optional research studies
We would like to collect additional blood to learn more about how chemotherapy drugs are broken down in your body, to see if certain genes (genes direct the activities of cells) will affect whether you experience bad effects after chemotherapy, whether your immune system is able to find neuroblastoma tumor cells and to test new methods for finding tumor cells. We will obtain all blood specimens through your central venous catheter. We will try to collect additional blood when you are already having blood drawn for routine purposes.
We would like to use any leftover portions of your bone marrow or stem cells that are not needed to treat you to carry out special biology research studies to learn more about neuroblastoma. These studies will test new methods for finding tumor cells.
Blood tests
Before starting treatment, we would like to take additional blood ( teaspoons) to see if your immune cells can recognize neuroblastoma tumors.
Before starting treatment, we would like to take additional blood ( ) to look at how certain genes (genes direct the activities of cells) may effect whether you have side effects after chemotherapy.
During Cycle 1 and Cycle 2 of Induction therapy we would like to take additional blood ( just less than 1 teaspoon) to study how topotecan is broken down by your body.
Before starting Induction therapy and at the end of Induction therapy, we would like to collect additional blood ( just less than 1 teaspoon) to test new methods of finding neuroblastoma.
Bone marrow Tests
We would also like to take of extra bone marrow at diagnosis and at the end of induction and to take 0.5 mL (1/10 of a tsp) after Cycle 2 of Induction to study new methods of finding tumor cells. This bone marrow will be taken when we are normally taking a sample to see how you are responding to therapy. The extra bone marrow will be shipped to a central lab for storage.
Peripheral Blood Stem Cell Tests
We would also like to obtain of stem cells that you do not need for your treatment to store for future research. These stem cells will be collected during your standard stem cell collection and will be shipped to a central lab for storage.
You and your physicians will not get the results of these blood, bone marrow and stem cell tests.. You will not benefit from allowing us to perform these extra studies. We will try to take the blood samples when we are already taking blood for standard therapy. The blood will be taken from your central line and will not require extra blood draws. You can request that we stop collecting these specimens at any time during your treatment. You will continue to get treatment on the study whether or not you allow us to collect these additional samples.
Please read each sentence below and think about your choice. After reading each sentence, choose "Yes" or "No" then add your initials and date after your answer. No matter what you decide to do, it will not affect your care. If you have any questions, please talk to your doctor or nurse, or call our research review board at IRB's phone number included in this consent.
I agree to have additional blood obtained for research purposes (to test for immune cells that recognize neuroblastoma).
Yes
No Initials
I agree to have additional blood obtained for research purposes (to test if certain genes are associated with side effects).
Yes
No Initials
I agree to have additional blood obtained for research purposes (to test how topotecan is broken down).
Yes No Initials
I agree to have additional blood obtained for research purposes (to find new ways to detect neuroblastoma).
Yes No Initials
I agree to have additional bone marrow obtained for research purposes (to find new ways to detect neuroblastoma)
Yes No Initials
I agree to have extra peripheral blood stem cells used for research purposes (to find new ways to detect neuroblastoma)
Yes
No Initials
HOW LONG WILL I BE ON THIS STUDY?
The first part of the treatment takes about 5 months. As long as your tumor doesn't get worse, you will be offered the option to continue on Part 2 of this clinical trial once you have completed Induction therapy. You will need to get more therapy for neuroblastoma whether or not you take part in Part 2.
We will continue to collect some medical information about how you are doing for 10 years after you enter the study. Keeping in touch with you and checking on how your health is every year for a while after you complete treatment helps us understand the long-term effects of the study.
Your doctor or the study doctor may decide to take you off this study for the following reasons:
he/she believes that it is your best interest
your disease comes back during treatment
you had side effects from the treatment that are considered too severe
new information becomes available that shows that another treatment would be better for you
You can stop participating at any time. However, if you decide to stop participating in the study, we encourage you to talk to the study doctor and your regular doctor first.
WHAT ARE THE RISKS OF THE STUDY AND HOW ARE THE RISKS DIFFERENT FROM TREATMENT?
Treatment Risks
All people who get cancer treatment are at risk of having side effects. In addition to killing tumor cells, cancer chemotherapy can damage normal tissue and produce side effects. Side effects are usually reversible when the medication is stopped but occasionally persist and cause serious complications. A person can die from these and other complications.
Common side effects include nausea, vomiting, hair loss, and fatigue. Drugs may be given to prevent or decrease nausea and vomiting. Hair loss is usually temporary but on very rare occasions it may be permanent. Some chemotherapy may lead to sterility. Sterility is the inability to have children. There is also the possibility that a second cancer may develop years later as a result of the chemotherapy. The risks of the individual drugs given as standard treatment are listed on the tables in Attachment #1. Side effects can be increased when chemotherapy drugs are combined.
The most common serious side effect from cancer treatment is lowering of the number of blood cells resulting in anemia, increased chance of infection, and bleeding tendency. Low blood counts are described in your Family Handbook for Children with Cancer. You will be taught more about caring for your child when his or her blood counts are low.
There is a risk that the treatment plan will not cure the cancer or that the cancer can go away after the treatment and then come back at a later date.
Reproductive risks:
Because the drugs and/or radiation therapy in this study can affect a developing fetus (unborn baby in the womb), you should not become pregnant or father a baby while on this study. Ask about counseling and more information about preventing pregnancy. The administration of chemotherapy described may cause infertility (being less able to produce a viable egg or sperm) or sterility (being unable to produce a viable egg or sperm). We will talk to males who have reached puberty about sperm banking.
For Women:
The treatment on this study can affect an unborn child. You should not become pregnant or breast feed your baby while being treated on this study. If you are sexually active and are at risk of getting pregnant, you and your male partner(s) must use an effective method to avoid pregnancy or you must not have sex. The study doctor will talk to you about acceptable methods to avoid pregnancy while you are being treated on this study. You will have to use the chosen method to avoid pregnancy or abstain (not have sexual intercourse) the whole time you are being treated on this study. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid pregnancy or abstain. Natural family planning and the rhythm method will not be permissible means of avoiding pregnancy during study participation. If you have questions about this or want to change your method to avoid pregnancy during therapy, please ask your doctor. If you
become pregnant during the research study, please tell the investigator and your doctor immediately.
If you are nursing a baby, the drugs used in this research could pass into the breast milk. You should not nurse your baby for the whole time you are getting the study medicines. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid nursing.
For Men:
The treatment on this study can damage sperm. You should not father a child while on this study as the treatment may indirectly affect an unborn child. If you are sexually active and are at risk of causing a pregnancy, you and your female partner(s) must use a method to avoid pregnancy that works well or you must not have sex. The investigator will talk to you about the acceptable methods to avoid pregnancy while you are being treated on this study. You will have to use the chosen method to avoid pregnancy or abstain (not have sexual intercourse) the whole time you are being treated on this study. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid pregnancy or abstain. Natural family planning and the rhythm method will not be permissible means of avoiding pregnancy during study participation. If you have questions about this or want to change your method to avoid pregnancy during therapy, please ask your doctor. If your partner becomes pregnant during the research study, please tell the investigator and your doctor immediately.
Risks of Study
The use of the topotecan/cyclophosphamide combination may lead to increased side effects compared to standard chemotherapy.
The use of topotecan/cyclophosphamide may not work as well as the current standard treatment.
In addition to the risks described above, there may be unknown risks, or risks that we did not anticipate, associated with being in this study.
Risks and side effects related to cyclophosphamide include those which are:
Likely
Less Likely
Rare But Serious
- Loss of appetite
- Nausea
- Vomiting
- Fewer white blood cells in the blood.
- A low number of white blood cells may make it easier to get infections.
- Hair loss
- Decreased ability of the body to fight infection
- Absence or decrease in the number of sperm which may be temporary or permanent which may decrease the ability to
- Abnormal hormone function which may lower the level of salt in the blood
- Abdominal pain
- Diarrhea
- Fewer red blood cells and platelets in the blood
- A low number of red blood cells may make you feel tired and weak.
- A low number of platelets may cause you to bruise and bleed more easily.
- Bleeding and
- Heart muscle damage which may occur with very high doses and which may be fatal
- Abnormal heart rhythms
- Damage and scarring of lung tissue which may make you short of breath
- A new cancer or leukemia resulting from this treatment.
- Damage or scarring of urinary bladder tissue
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart
have children
inflammation of the urinary bladder
- Absence or decrease monthly periods which may be temporary or permanent and which may decrease the ability to have children
- Temporary blurred vision
- Nasal stuffiness with IV infusions
- Skin rash
- Darkening of areas of the skin and finger nails
- Slow healing of wounds
- Infections
rate chills and fever
- Infertility which is the inability to have children
Risks and side effects related to topotecan include those which are:
Likely
Less Likely
Rare But Serious
- Diarrhea
- Nausea
- Vomiting
- Constipation
- Fewer white blood cells, red blood cells and platelets in the blood.
- A low number of white blood cells can make it easier to get infections
- A low number of red blood cells can make you feel tired and weak
- A low number of platelets causes you to bruise and bleed more easily
- Fever including fever with a low white blood cell count which could indicate infection and may require hospitalization and treatment with antibiotics
- Pain which may be in your abdomen, back or bones
- A feeling of weakness and/or tiredness
- Loss of appetite
- Elevation in the blood of certain enzymes or bilirubin found in the liver which could indicate liver irritation or damage
- Headache
- Rash, hives, itching or a red bumpy rash
- A mild lowering of the blood pressure which usually does not require treatment
- Inflammation and/or sores in the mouth, throat and/or esophagus
- An infection in the blood which will require admission to the hospital and treatment with antibiotics
- Numbness and tingling in the fingers and toes
- Small amount of blood and/or protein in the urine or an elevation in blood creatinine which may indicate mild kidney damage
- Shortness of breath
- Muscle or joint aches
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure and a rapid heart rate
- Severe allergic reaction which can be life threatening with rapid build-up of fluid under the skin, in the lining of the intestine and possibly in the throat or swelling of the tongue which could make it difficult to breath.
- Bleeding into the tumor which may cause damage depending on the location of the tumor
Children's Oncology Group
Temporary hair loss
and pains
Chest pain
Shaking chills
ARE THERE BENEFITS TO TAKING PART IN THE STUDY?
We hope that you will get personal medical benefit from participation in this clinical trial, but we cannot be sure that will happen. These potential benefits could include better chance of a cure or less side effects.
We expect that the information learned from this study will benefit other patients in the future.
WHAT OTHER OPTIONS ARE THERE?
Instead of being in this study, you have these options:
Current standard therapy even if you do not take part in the study. Standard therapy is described on page 2.
Taking part in another study
You are encouraged to discuss these options with your regular doctor as well as other trusted personal and family advisors.
WHAT ABOUT CONFIDENTIALITY?
Efforts will be made to keep your personal information confidential. We cannot guarantee absolute confidentiality. Your personal information may be disclosed if required by law.
The Children's Oncology Group has received a Certificate of Confidentiality from the federal government, which will help us protect the privacy of our research subjects. Information about the certificate is attached at end of this consent.
Organizations that may inspect and/or copy your research records for quality assurance and data analysis include groups such as:
The Children's Oncology Group
Representatives of the National Cancer Institute (NCI), Food and Drug Administration (FDA), and other U.S. and international governmental regulatory agencies involved in overseeing research
The Institutional Review Board (IRB) of this hospital
Pediatric Central Institutional Review Board (CIRB) of the National Cancer Institute
WHAT ARE THE COSTS?
Taking part in this study may lead to added costs to you or your insurance company. Please ask about any expected added costs or insurance problems. Staff will be able to assist you with this.
You or your insurance company will not be charged for research studies performed on blood, bone marrow or stem cells.
In the case of injury or illness resulting from this study, emergency medical treatment is available but will be provided at the usual charge. No funds have been set aside to compensate you in the event of injury. However by signing this form, you are not giving up any legal rights to seek to obtain compensation for injury.
You or your insurance company will be charged for continuing medical care and/or hospitalization.
For more information on clinical trials and insurance coverage, you can visit the National Cancer Institute's Web site at http://cancer.gov/clinicaltrials/understanding/insurance-coverage. You can print a copy of the "Clinical Trials and Insurance Coverage" information from this Web site.
There are no plans to pay you for taking part in this study. If this study includes providing specimens to the researcher, there is no intention or plan for you to profit from any new products developed from research done on your specimens.
WHAT ARE MY RIGHTS AS A PARTICIPANT?
Taking part in this study is voluntary. You may leave the study at any time. Refusing to take part in the study, or leaving the study will not result in any penalty or loss of benefits to which you are entitled. Your doctor will still take care of you.
We will tell you about new information that may affect your health, welfare, or willingness to stay in this study. A committee outside of COG closely monitors study reports and notifies institutions if changes must be made to the study. Members of COG meet twice a year to evaluate results of treatment and to plan new treatments.
During your follow-up visits after treatment, you may ask to be given a summary of the study results after they are written up. This may be several years after treatment for all people on the study is completed.
WHOM DO I CALL IF I HAVE QUESTIONS OR PROBLEMS?
For questions about the study or a research-related problem, or if you think you have been injured, you may contact Dr. XXXX or your doctor at XXXXX.
If you have any questions about your rights as a research participant or any problems that you feel you cannot discuss with the investigators, you may call XXXX IRB Administrator at (XXXX
If you have any questions or concerns that you feel you would like to discuss with someone who is not on the research team, you may also call the Patient Advocate at XXXX
WHERE CAN I GET MORE INFORMATION?
The COG Family Handbook for Children with Cancer has information about specific cancers, tests, treatment side effects and their management, adjusting to cancer, and resources.
A description of this clinical trial will be available at: http://www.ClinicalTrials.gov, as required by U.S. Law. This web site will not include information that can identify you. At most, the web site will include a summary of the results. You can search this web site at any time.
You will get a copy of this form. You may also ask for a copy of the protocol (full study plan).
SIGNATURE
I have been given a copy of all [insert total number of pages] pages of this form.
The form includes three (3) attachments.
I agree to take part in this study.
Participant Date
Parent/Guardian Date
Parent/Guardian Date
Physician/PNP obtaining consent Date IRB#
IRB Approved:
Attachment #1
Risks of individuals drugs given as standard treatment for Neuroblastoma
Risks and side effects related to cisplatin include those which are:
Likely
Less Likely
Rare But Serious
- Nausea and vomiting
- fewer red blood cells and white blood cells and platelets in the blood
- a low number of red blood cells can make you feel tired and weak
- a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and bleed more easily
- Abnormal levels of magnesium in the body which may require that you take extra magnesium by mouth or in the vein
- Loss of appetite
- Damage to the ear causing difficulty in hearing high pitched sounds
- Temporary and mild increases in levels of certain chemicals in the blood because the kidney is not working as well as normal
- Abnormal levels of certain salts in the body like sodium, calcium, potassium and phosphate
- Metallic taste
- Rash
- Numbness and tingling in the fingers and toes
- Temporary changes in vision
- Damage to the ear causing hearing loss, balance problems and ringing in the ears
- Elevation in the blood of certain enzymes found in the liver which may indicate liver irritation or damage.
- Inflammation and discomfort in the vein through which the medicine was given
- Damage to the skin may occur if the medication leaks from the vein
- Allergic reactions which may be severe and lifethreatening, causing difficulty in breathing, rapid heart rate, facial swelling and or a drop in blood pressure
- Damage to the kidney which may be permanent
- Deafness
- Seizures
- Damage to the vision which could lead to blurred vision, bluegreen color blindness and to loss of vision which usually goes away after stopping the drug.
- Decrease in muscle and nerve reflexes that may affect normal functions such as walking
- Leukemia later in life
Risks and side effects related to doxorubicin include those which are:
Likely
Less Likely
Rare But Serious
- Nausea
- Vomiting
- Temporary hair loss
- Pink or red color to urine, sweat, tears, saliva
- Fewer white blood cells, red blood cells and platelets in the blood.
- A low number of red blood cells can make you feel tired and weak
- A low number of white blood cells can make it easier to get infections
- A low number of platelets causes you to bruise and bleed more easily
- Slight damage to the heart muscle that is unlikely to have any noticeable effects on your heart function
- Inflammation and/or sores in the mouth (and/or throat and /or esophagus, the tube that leads from the mouth to the stomach) that may make swallowing difficult and are painful (painful mouth sores)
- Damage to the heart muscle which may make you tired, weak, feel short of breath, and retain fluid
- Facial Flushing
- Fever/chills
- Hives
- High levels of uric acid in the blood which could damage the kidneys
- Dark discoloration of the hands, feet and under the fingernails with possible separation of the nail from the nail bed.
- Damage to the skin if the medication leaks from a vein
- Thickening and hardening of the veins through which the medication is given
- Reddening reaction of the vein through which the drug is given.
- Elevation in the blood of certain enzymes found in the liver which may indicate liver irritation or damage.
- Tearing and inflammation of the eyes
- Loss of appetite
- Redness and burning at sites which have received radiation in the past
- Diarrhea
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure and a rapid heart rate
- Ulceration of the lower intestinal tract
- An irregular heart beat which can be lifethreatening
- Severe damage to the heart muscle which may lead to severe heart failure
- A new cancer or leukemia resulting from this treatment.
The risk of heart damage may be greater in very young children than in older ones
Risks and side effects related to etoposide include those which are:
Likely
Less Likely
Rare But Serious
- Nausea and vomiting
- Hair Loss
- A feeling of weakness or tiredness
- fewer red and white blood cells and platelets in the blood
- a low number of red blood cells can make you feel tired and weak
- a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and bleed more easily
- Loss of appetite
- Decreased blood pressure during the infusion which may require treatment
- rashes
- Diarrhea
- Pain in the abdomen
- Mouth sores
- Tingling sensation or loss of sensation in fingers or toes
- A feeling of extreme tiredness or weakness
- The finger or toe nails may loosen from their nail beds
- Inflammation of the vein through which the medication was given
- Chest pain
- Damage to the liver
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate chills and fever
- A new cancer or leukemia resulting from this treatment
- Severe rashes which can result in loss of skin and damage to mucous membranes
- Absence or decrease monthly periods which may be temporary or permanent and which may decrease the ability to have children
- Damage to the heart muscle which may make you feel tired, weak, feel short of breath, and retain fluid
Risks and side effects related to myeloid growth factors (filgrastim (G-CSF) or pegfilgrastim) include those which are:
Likely
Less Likely
Rare but serious
- Aching or pain in the bones
- Pain, redness, itching, and hardening of the skin and bruising at the site of the injection
- Headache
- Higher than normal levels of liver enzymes in the blood which may indicate liver irritation or damage
- Increase of uric acid in the blood
- A low number of platelets in the blood which may cause you to bruise and bleed more easily
- Low fever
- Enlargement of the spleen (an organ in the abdomen/belly which stores blood cells)
- Allergic reactions which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, hives, itching, and facial swelling.
- Serious allergic reaction which can be life threatening with rapid build-up of fluid under the skin, in the lining of the intestine, and possibly in the throat or swelling of the tongue which could make it difficult to breath
- If you are known to have sickle cell disease, filgrastim or pegfilgrastim may cause a sickle cell crisis.
which may cause pain in the abdomen or left shoulder
- Higher than normal white blood count
- Skin condition marked by fever and painful skin lesions that appear mainly on the face, neck, back and arms
- Rash or worsening of rash
- Inflammation of blood vessels in the skin leading to a raised purple rash and bruising has been seen mainly in patient who are treated for a long time
- Overall reddening with feelings of warmth
- Severe damage to the spleen (an organ in the abdomen/belly which stores blood cells) which could lead to pain and loss of blood into the abdomen (belly) and maybe life threatening
- Difficulty breathing and lung damage that may be due to the white blood cells that are stimulated by filgrastim or pegfilgrastim traveling to the lungs when they are inflamed or infected.
- A blood disorder or leukemia that has only been seen in patients with certain immune disorders who are treated for a very long time
Reported with filgrastim Reported with pegfilgrastim
Risks and side effects related to mesna IV include those which are:
Likely
Less Likely
Rare But Serious
- Nausea.
- Vomiting
- Stomach pain.
- Headache.
- Pain in arms, legs and joints.
- Tired feeling.
- Rash.
- Temporary low blood pressure.
- Diarrhea.
- Fever
- Facial flushing with red cheeks
- Nervousness
- Dizziness
- Confusion
- Swelling around the eyes
- Coughing
- Rapid heart rate
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate chills and fever
Risks and side effects related to vincristine include those which are:
Likely
Less Likely
Rare But Serious
- Hair loss
- Reversible nerve problem that may affect the way you walk or the feelings in your fingers or toes
- Constipation
- Jaw pain
- Headache
- Muscle weakness
- Pain and bloating in your abdomen
- Numbness and tingling
- Wrist or foot drop
- Drooping eyelids
- Double vision, difficulty seeing at night
- Hoarseness of your voice
- Difficulty sweating
- Abnormal walk with foot slapping
- Difficulty with urination or increase desire to urinate
- Dizziness and low blood pressure when you stand
- Abnormal hormone function which may lower the level of salt in the blood
- A mild drop in white blood cells, red blood cells and platelets in the blood
- a low number of red blood cells can make you feel tired and weak
- a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and bleed more easily
- Complete stoppage of your intestinal activity which can result in intestinal blockage
- If the drug leaks out of the vein when being administered it will cause damage to nearby tissue
- Seizures
- Vocal cord paralysis
- Difficulty breathing
- Inability to walk
- Decreased ability to hear clearly
- Damage to the nerve to the eye (optic nerve) leading to decreased vision and possible blindness
- In combination with other chemotherapy drugs: damage to the liver which can lead to inflammation and/or scarring which could lead to a yellow appearing skin, and fluid collection in the abdomen (belly) which makes it look larger
These procedures are usually safe. Side effects that can occur during PBSC collection include nausea, vomiting, fainting or dizziness, seizures, skin rash, hives, flushing (redness and warmness of the skin, usually the face), blood loss, and infection. Tingling of the lips, muscle cramping and, very rarely, changes in the heart rhythm can occur. These can be prevented or made milder by giving calcium supplements, either by mouth or IV. Very rarely, (less than 1 in 1,000 procedures), clotting may occur in the apheresis machine or in a patient and is potentially life-threatening. To reduce the risk of clotting, you will be given a drug called ACD (acid-citratedextrose). This drug may increase the risk of bleeding and may cause temporary tingling of the lips and limbs, muscle cramping, seizures, or changes in the heart rhythm.
Attachment #3
Certificate of Confidentiality Information
The Children's Oncology Group has received a Certificate of Confidentiality from the federal government, which will help us protect the privacy of our research subjects. The Certificate protects against the involuntary release of information about subjects collected during the course of our covered studies. The researchers involved in the studies cannot be forced to disclose the identity or any information collected in the study in any legal proceedings at the federal, state, or local level, regardless of whether they are criminal, administrative, or legislative proceedings. However, the subject or the researcher may choose to voluntarily disclose the protected information under certain circumstances. For example, if the subject or his/her guardian requests the release of information in writing, the Certificate does not protect against that voluntary disclosure. Furthermore, federal agencies may review our records under limited circumstances, such as a DHHS request for information for an audit or program evaluation or an FDA request under the Food, Drug and Cosmetics Act. The Certificate of Confidentiality will not protect against the required reporting by hospital staff of information on suspected child abuse, reportable communicable diseases, and/or possible threat of harm to self or others.
SAMPLE INFORMED CONSENT / PARENTAL PERMISSION FOR PARTICIPATION IN RESEARCH
This model informed consent form has been reviewed by the DCT/NCI and is the official consent document for this study. Local IRB changes to this document are allowed. (Institutions should attempt to use sections of this document which are in bold type in their entirety.) Editorial changes to these sections may be made as long as they do not change information or intent. If the local IRB insists on making deletions or more substantive modifications to the risks or alternatives sections, they must be justified in writing by the investigator and approved by the IRB .
ANBL0532, Phase III Randomized Trial of Single vs. Tandem Myeloablative Consolidation Therapy for High-Risk Neuroblastoma:
PART 2 A: CONSENT FOR PATIENTS RANDOMIZED TO CONSOLIDATION THERAPY
Consolidation and Maintenance Phases of Therapy for High Risk Neuroblastoma (Please note that this consent MUST be signed before the patient proceeds for consolidation therapy)
When the word "you" appears in this consent form, it refers to you or your son or daughter; "we" means the doctors and other staff.
WHY ARE YOU BEING INVITED TO TAKE PART IN THIS STUDY?
This is a clinical trial, a type of research study. Your study doctor will explain the clinical trial to you.
The Children's Oncology Group (COG) is carrying out this study. COG is an international research group that conducts clinical trials for children with cancer at more than 200 hospitals in the United States, Canada, Australia, New Zealand, the Netherlands, and Switzerland. It is common medical practice to treat children with cancer on research studies like this one.
You are being asked to take part in this study because you have high risk neuroblastoma and you have participated in Part 1 of this study. Neuroblastoma is a type of cancer. You have the type of neuroblastoma that is called High Risk because your tumor has spread from where it started from or because your type of tumor is harder to treat.
It is common to enroll children and adolescents with cancer in a clinical trial that seeks to improve cancer treatment over time. Clinical trials include only people who choose to take part. You have a choice between a standard treatment for High Risk neuroblastoma and this clinical trial. Please take your time to make your decision about taking part in this clinical trial. You may discuss your decision with your friends and family. You can also discuss it with your health care team. We encourage you to include your child in the discussion and decision to the extent that she or he is able to understand and take part. If you have any questions, you can ask your study doctor for more explanation.
WHAT IS THE CURRENT STANDARD OF TREATMENT FOR THIS DISEASE?
The treatment for neuroblastoma includes 3 parts (phases) of therapy called Induction, Consolidation and Maintenance. During Induction therapy anti-cancer drugs (chemotherapy)
and surgery are used to kill and remove as much tumor as possible. Blood stem cells are collected during the Induction phase of therapy. After collection the blood stem cells are frozen and stored to be used during Consolidation phase of treatment. Blood stem cells are the cells that create new blood cells, such as red blood cells, white blood cells, and platelets. Blood stem cells can be collected from the blood by using a machine that can separate out the part of blood that has stem cells and then return the remaining blood back to the patient. You have already received this part of therapy.
During the Consolidation phase of treatment extremely high doses of chemotherapy are given to better kill any remaining neuroblastoma cells. It is standard therapy to give one cycle of this extremely high dose chemotherapy. The high doses of chemotherapy destroy healthy bone marrow. Bone marrow is the soft tissue in the hollow of flat bones of the body that produces new blood cells. The peripheral blood stem cells that were stored during the Induction phase of treatment are given back to the patient after the high dose Consolidation chemotherapy. These stems cells allow the bone marrow to return to normal so that new blood cells can be made. This type of therapy is called a Hematopoietic Stem Cell Transplant. Once the patient has healed from the effects of high doses of chemotherapy, radiation therapy is given to the first place the tumor was found and to any additional places where the tumor was found after Induction therapy.
During the third phase of therapy, Maintenance, an oral drug, called cis-retinoic acid (Accutane) is given.
WHY IS THIS STUDY BEING DONE?
The main goal of this research study is to find out if using 2 cycles of high dose Consolidation chemotherapy, instead of 1 cycle, will decrease the chance that the neuroblastoma will grow back after therapy. In this study, you will get either 2 cycles of high dose Consolidation chemotherapy or 1 cycle of high dose Consolidation chemotherapy. It is standard to give only one cycle of high dose Consolidation chemotherapy. Smaller clinical trials have studied the safety of giving 2 cycles of high dose Consolidation chemotherapy to treat high risk neuroblastoma. We do not know whether using 2 cycles of Consolidation chemotherapy will be better for treatment of high risk neuroblastoma.
Tandem stem cell transplant for children with neuroblastoma is experimental.
This research study will also find out if patients whose tumor did not disappear with initial induction therapy will need a higher dose of radiation therapy.
This study is being done because more than half of the patients with high risk neuroblastoma will not be cured of their disease.
The treatment is described in two separate consent forms. You have already read the first consent form (Part 1) and completed Part 1 of treatment. We are now presenting Part 2 (the second consent form) of the study. The second consent (Part 2) will discuss the Consolidation and Maintenance phases of therapy.
During this part of the study, we will:
Compare the effects, good and/or bad, of using 2 cycles of high dose Consolidation chemotherapy with 1 cycle of high dose Consolidation chemotherapy on children with high risk neuroblastoma to find out which is better.
Study whether a higher dose of radiation, given only to patients with tumor remaining
after induction therapy, will decrease the chance that tumor will grow back.
Study whether your immune system (blood cells that help fight infection) can produce cells that can recognize and kill neuroblastoma.
Study whether new tests can be used to find small amounts of neuroblastoma cells.
Study how cis-retinoic acid (Accutane) is broken down by your body and study whether your genes (genes direct activities of cells) effect how you respond to cis-retinoic acid therapy.
HOW MANY PEOPLE WILL TAKE PART IN THIS STUDY?
About 664 subjects will take part in this study. The study will last about 3-4 years.
WHAT WILL HAPPEN ON THIS STUDY?
Random Assignment
If you participate in this part of the study you will be given one of 2 different treatment plans. The treatment plan you get is decided by a process called randomization. Randomization means that the treatment is assigned based on chance. It is a lot like flipping a coin, except that it is done by computer to make sure that there are about the same number of people on each treatment plan of the study. You or your doctor can not choose what treatment you will have.
Treatment Plan
The treatment plan involves cancer fighting medicine called chemotherapy plus radiation therapy. The treatment on this part of the clinical trial takes about 9-11 months. It is divided into 2 stages.
The two different treatment plans are the same except for some differences during the Consolidation phase of therapy. The rest of the treatment that is given is standard therapy for people with high risk neuroblastoma.
The two treatment arms are as follows:
A: Single Myeloablative Consolidation Therapy
B: Tandem Myeloablative Consolidation Therapy
Myeloablative therapy means that you will receive very high doses of chemotherapy that will destroy bone marrow cells. Stem cells are given back after the chemotherapy to replace the bone marrow cells that were destroyed.
You will receive consolidation treatment after you have finished induction therapy and signed this consent.
Randomization
After you have finished induction therapy and signed this consent, you will be randomized to receive either a single (one cycle) of extremely high dose chemotherapy (Single Myeloablative Consolidation Therapy) or tandem (two cycles) of extremely high dose chemotherapy (Tandem Myeloablative Consolidation Therapy).
The rest of the Consolidation therapy and Maintenance therapy will be the same in both groups of patients.
Diagram of treatment
A diagram of part 2 of the treatment can be seen below.
Children's Oncology Group
Treatment Plan Tables
The following drug therapies compare standard consolidation chemotherapy and stem cell transplant to the experimental tandem consolidation chemotherapy and transplant.
Various methods will be used to give drugs:.
PO - Drug is given by tablet or liquid swallowed through the mouth (PO).
IV - Drug is given using a needle or tubing inserted into a vein. It can be given by IV push over several minutes or by infusion over minutes or hours
SUBQ - Drug is given by injecting a needle into the tissue just under the skin (SUBQ shot)
Most drugs on this study will be given using a needle or tubing inserted into a vein (IV).
Treatment for participants who are on treatment plan A
After you finish induction therapy, you will get one cycle of very high dose chemotherapy and a hematopoietic stem cell transplant. The transplant will begin with treatment with very high doses of the chemotherapy drugs carboplatin, etoposide and melphalan. Etoposide and carboplatin are given as a continuous infusion for 4 consecutive days. The drug melphalan is given as a daily dose for 3 days. After the high dose chemotherapy treatment, your stored stem cells will be given IV (through the central line). These stem cells were collected from you during the induction phase of therapy and frozen. This is called a Hematopoietic Stem Cell Transplant. You will be given filgrastim (GCSF) starting on the same day you receive your stem cells and continuing until your white blood cells have returned to a safe number, usually about 2 weeks.
Drug
How the drug will be given
Days
Carboplatin
IV over 24 hours
Etoposide
IV over 24 hours
Melphalan
IV over 15-30 minutes
Blood Stem Cells
IV
0
Filgrastim (GCSF)
SUBQ (or) IV
Treatment for participants who are on treatment plan B
After completion of induction therapy, you will begin the first of the two cycles of very high dose chemotherapy and hematopoietic stem cell transplant. The first transplant will begin with treatment with very high doses of the chemotherapy drugs thiotepa and cyclophosphamide given IV over 6 total days. After the high dose chemotherapy treatment, some of your stored stem cells will be given IV (through the central line). These stem cells were previously collected from you during the induction phase and frozen. You will be given filgrastim (GCSF) starting on the same day you get your stem cells and you will continue to get filgrastim (GCSF) until your white blood cells have returned to a safe number, usually about 2 weeks.
Drug
How the drug will be given
Days
Thiotepa
IV over 2 hours
Cyclophosphamide
IV over 1 hour
Mesna
IV over 15 minutes
Blood Stem Cells
IV
0
GCSF
SUBQ (or) IV
Between 6-10 weeks from the start of the first treatment a second cycle of very high dose treatment is given. The treatment includes the drugs etoposide and carboplatin, which are given as a continuous infusion for 4 consecutive days. The drug melphalan is also given as a daily dose for 3 days during this treatment. After this second high dose chemotherapy treatment, some of your remaining stored stem cells will be given back to you. You will be given the same care as the first transplant. You will be given filgrastim (GCSF) starting on the same day you get your stem cells and you will continue to get filgrastim (GCSF) until your white blood cells have returned to a safe number, usually about 2 weeks.
Drug
How the drug will be given
Days
Carboplatin
IV over 24 hours
Etoposide
IV over 24 hours
Melphalan
IV over minutes
Blood Stem Cells
IV
0
Filgrastim (GCSF)
SUBQ (or) IV
Radiation Therapy
It is standard to give radiation treatment to the area of the main tumor and to areas that still showed signs of active disease at the end of Induction phase of treatment. Radiation therapy will begin after you have gotten better from the immediate side effects of the high doses of chemotherapy. If you are randomized to arm B (2 cycles of consolidation chemotherapy) you will get radiation therapy after you have recovered from the transplant. Radiation therapy usually starts about a month after the transplant. The radiation therapy doctors will discuss this with you in more detail. The radiation is given in one short session each day, for a period of about 3-4 weeks. Radiation to the stomach may cause nausea and vomiting, and often causes the blood counts to drop temporarily, which may require treatment with filgrastim (GCSF) or transfusions (give you blood). Other risks are discussed below.
You will get a higher dose of radiation treatment if x-ray tests performed at the end of Induction therapy still show tumor (a lump). The higher dose of radiation will only be given to the area where tumor is present. This higher dose of radiation is experimental. We are studying whether this higher dose of radiation will decrease the risk of tumor growing back.
MAINTENANCE PHASE OF THERAPY
Maintenance therapy will begin after you have recovered from effects of radiation therapy. The use of an immune therapy that targets neuroblastoma (a medicine called chimeric 14.18 antibody) combined with the prior standard therapy of cis-Retinoic acid (Accutane) has recently been shown to improve survival for children with high risk neuroblastoma. Chimeric 14.18 antibody is only available to patients with high risk neuroblastoma by enrolling onto another clinical trial. It is strongly encouraged that you speak with your physician about receiving chimeric 14.18 therapy. If you are not able to get ch14.18 therapy or choose not to get it, then you will get cis-Retinoic acid (Accutane) therapy alone. Cis-Retinoic acid is given by mouth, twice daily for 2 weeks, followed by two weeks without the drug. This cycle will be repeated 6 times for a total of 6 months of treatment.
Standard Medical Tests
Before treatment on this study begins, and while receiving treatment, you will have a series of standard medical tests:
Physical exam
Blood Tests
Bone marrow tests
Tests of vision and hearing
Urine tests
Various scans (x-ray tests to see where the tumor is in your body)
Tests of kidney function
Tests of lung and heart function
Hearing tests
Dental exams
Research study tests and procedures
The following tests will be done because you are part of this study. These tests are not part of standard care.
Some copies of the scans (x-rays) used to make the diagnosis of your disease and to see how your tumor responded to therapy will be sent to a central review center as part of COG quality control.
We would like to collect additional blood to see if certain genes (genes direct the activities of cells) will effect whether you experience bad effects after chemotherapy, to see if your immune system is able to recognize neuroblastoma tumor cells and to test new methods for finding tumor cells. We will get all blood specimens through your central venous catheter. We will try to get these additional blood samples when you are already having blood drawn for routine purposes.
We would like to use any leftover portions of your bone marrow that is not needed to treat you to carry out special biology research studies to learn more about neuroblastoma. These studies will test new methods for finding tumor cells.
Blood tests
Before starting cis-retinoic acid (Accutane) treatment, we would like to take additional blood (20 teaspoons) to see if your immune cells can recognize neuroblastoma tumors.
After you have received the first 14 days of cis-retinoic acid, we would like to take additional blood ( ) to look at how certain genes (genes direct the activities of cells) may affect whether you have side effects.
At the end of Consolidation phase of therapy (after completing radiation therapy) and at the end of Maintenance therapy, we would like to obtain additional blood ( teaspoon) to test new methods of finding neuroblastoma.
Bone marrow tests
We would also like to take 0.5 ml (1/10 tsp) of extra bone marrow at the end of Consolidation therapy and at the end of Maintenance therapy to study new methods of finding tumor cells. This bone marrow will be taken when we are normally taking a sample to see how you are responding to therapy. The extra bone marrow will be shipped to a central lab for storage.
You and your physicians will not be given the results of these blood and bone marrow tests. You will not benefit from allowing us to perform these extra studies. We will try to take the blood samples when we are already taking blood for standard therapy. The blood will be taken from your central line and will not require an extra blood draw. You can ask that we stop collecting these specimens at any time during your treatment. You will continue to get treatment on the study whether or not you allow us to collect these additional samples.
Please read each sentence below and think about your choice. After reading each sentence, choose "Yes" or "No" then add your initials and date after your answer. No matter what you decide to do, it will not affect your care. If you have any questions, please talk to your doctor or nurse, or call our research review board at IRB's phone number included in this consent.
I agree to the have additional bone marrow obtained for research purposes (to find new ways to detect neuroblastoma).
Yes No Initials
I agree to have additional blood obtained for research purposes (to test for immune cells that recognize neuroblastoma).
Yes No Initials
I agree to have additional blood obtained for research purposes (to find new ways to detect neuroblastoma).
Yes No Initials
I agree to have additional blood obtained for research purposes (to test how cis-RA is broken down).
Yes No Initials
HOW LONG WILL I BE ON THIS STUDY?
The Consolidation phase of therapy takes about 2-3 months (Arm A) or about 4-5 months (Arm B). The Maintenance phase of therapy will last an additional 6 months.
We will continue to collect some medical information about how you are doing for 10 years after you enter the study. Keeping in touch with you and checking on how your health is every year for a while after you complete treatment helps us understand the long-term effects of the study.
Your doctor or the study doctor may decide to take you off this study for the following reasons:
he/she believes that it is your best interest
your disease comes back during treatment
you experiences side effects from the treatment that are considered too severe
new information becomes available that shows that another treatment would be better for you
You can stop participating at any time. However, if you decide to stop participating in the study, we encourage you to talk to the study doctor and your regular doctor first.
WHAT ARE THE RISKS OF THE STUDY?
Treatment Risks
All people who have cancer treatment are at risk of having side effects. In addition to killing tumor cells, cancer chemotherapy can damage normal tissue and produce side effects. Side effects are usually reversible when the medication is stopped but occasionally persist and cause serious complications. A person can die from these and other complications.
Common side effects include nausea, vomiting, hair loss, and fatigue. Drugs may be given to prevent or decrease nausea and vomiting. Hair loss is usually temporary but on very rare occasions it may be permanent. Some chemotherapy may lead to sterility. Sterility means that you will not be able to have children. There is also the possibility that a second cancer may develop years later as a result of the chemotherapy. The risks of the individual drugs given as standard treatment are listed on the tables in Attachment #1. Side effects can be increased when chemotherapy drugs are combined.
The most common serious side effect from cancer treatment is lowering of the number of blood cells resulting in anemia, increased chance of infection, and bleeding tendency. Low blood counts are described in your Family Handbook for Children with Cancer. The high doses of chemotherapy used during the Consolidation phase of treatment will cause very severe lowering of the blood cell numbers. You will be at risk to develop a life-threatening infection and bleeding following the very high doses of chemotherapy. To limit the risk of infection, your child will remain in a special hospital room until blood counts return to safe levels. Your stem cells that were previously collected and stored will be used to help your body make new blood cells.
It is possible that it will take a very long time for your blood cells to be made. This can happen if the stem cells were injured in your body even before they were removed or if the stem cells are injured during the collection, freezing or thawing processes. If this happens, you will have a high chance that you will develop a severe infection and/or bleeding. This is very unlikely to happen, but would almost certainly be fatal. Even after your body makes blood cells after the transplant, your ability to fight infection will be low for weeks to months after the transplant is complete. You will be more likely to develop an infection in the months after transplant, especially from viruses.
There is also a small risk of severe organ damage, especially to liver and kidney, with an even smaller risk to the heart and lungs. Though rare, the kidney damage can be severe enough to require dialysis. The liver damage can also be severe, resulting in jaundice and, occasionally, complete failure of the liver to function. This damage may be reversible or irreversible, and if irreversible, the liver failure would be fatal.
There is a chance that the stem cells may contain tumor cells. These cells could result in the tumor coming back.
These problems alone or in combination may be severe enough to be life-threatening. The purpose of using stem cells is to decrease the chance that these problems will happen. Your
doctor will also give you antibiotic and blood transfusions to decrease risk for infection or bleeding.
Months or years after treatment other side effects may appear.
We know that the chemotherapy drugs cyclophosphamide, melphalan and etoposide can cause leukemia.
Radiation therapy may cause bone cancers or other kinds of sarcomas.
Cyclophosphamide and melphalan will cause damage to the sexual glands. As a result, if you are a boy, you will almost certainly not be able to have biologically related children, and, if you are girl, you are likely to need hormones or other interventions to have children.
Reproductive risks:
It is unknown what effect(s) these treatments may have on an unborn child. As the drugs and/or radiation therapy in this study can affect a developing fetus (unborn baby in the womb), you should not become pregnant or father a baby while on this study. For this reason, if you are of child-bearing age, you will be asked to practice an effective method of birth control while participating on this study. The use of Isotretinoin can cause birth defects to unborn children if taken during pregnancy. Ask about counseling and more information about preventing pregnancy. Having the chemotherapy described may cause infertility (being less able to produce a viable egg or sperm) or sterility (being unable to produce a viable egg or sperm). We will talk to males who have reached puberty about sperm banking.
For Women:
The treatment on this study can affect an unborn child. You should not become pregnant or breast feed your baby while being treated on this study. If you are sexually active and are at risk of getting pregnant, you and your male partner(s) must use an effective method to avoid pregnancy or you must not have sex. The study doctor will talk to you about acceptable methods to avoid pregnancy while you are being treated on this study. You will have to use the chosen method to avoid pregnancy or abstain (not have sexual intercourse) the whole time you are being treated on this study. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid pregnancy or abstain. Natural family planning and the rhythm method will not be permissible means of avoiding pregnancy during study participation. If you have questions about this or want to change your method to avoid pregnancy during therapy, please ask your doctor. If you become pregnant during the research study, please tell the investigator and your doctor immediately.
If you are nursing a baby, the drugs used in this research could pass into the breast milk. You should not nurse your baby for the whole time you are getting the study medicines. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid nursing.
For Men:
The treatment on this study can damage sperm. You should not father a child while on this study as the treatment may indirectly affect an unborn child. If you are sexually active and are at risk of causing a pregnancy, you and your female partner(s) must use a method to avoid pregnancy that works well or you must not have sex. The investigator will talk to you about the acceptable methods to avoid pregnancy while you are being treated on this study. You will have to use the chosen method to avoid pregnancy or abstain (not have sexual intercourse) the whole time you are being treated on this study. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid pregnancy or abstain. Natural family planning and the rhythm method will not be
permissible means of avoiding pregnancy during study participation. If you have questions about this or want to change your method to avoid pregnancy during therapy, please ask your doctor. If your partner becomes pregnant during the research study, please tell the investigator and your doctor immediately.
There is a risk that the treatment plan will not cure the cancer or that the cancer can go away after the treatment and then come back at a later date.
Risks of Study
Tandem transplants are experimental. We have over 10 years of experience with tandem transplant, but we are still learning about the risks that happen after this type of treatment. The side effects described above may be increased if you have tandem ( 2 cycles) of high dose Consolidation chemotherapy.
If you receive the tandem therapy ( 2 cycles) you will receive an additional cycle of high dose Consolidation chemotherapy, cyclophosphamide and thiotepa. These drugs are often used for high dose Consolidation chemotherapy. You will receive them together, as the additional cycle of high dose Consolidation chemotherapy.
You may get a higher dose of radiation if you still had tumor remaining after finishing the Induction phase of treatment. For most patients, the area receiving the additional radiation dose will be small. However, it is possible that the use of higher doses of radiation may be more likely to cause the side effects of radiation that are described in Attachment #2.
Please see the attachment at the end of the consent (Attachment #2) for more information regarding the risks associated with radiation therapy and Peripheral Blood Stem Cell Harvesting and Reinfusion.
Information may also be found in the COG Family Handbook
For more information about risks and side effects, ask your child's study doctor.
In addition to the risks described above, there may be unknown risks, or risks that we did not anticipate, associated with being in this study.
Your physician will be checking closely to see if any of these side effects are occurring. Routine physical exams and laboratory tests will be done to monitor the effects of treatment. Side effects usually disappear after the treatment is completed.
Treatment on this study may have risks we don't know about. We may stop this treatment if we learn of serious, unexpected risks. We will explain the effects of stopping, and we will offer other treatments.
Risks and side effects related to the thiotepa include those which are:
Likely
Less Likely
Rare But Serious
- Nausea
- Pain at the injection site
- Severe allergic
- Vomiting
- Dizziness
reaction which can
- Loss of appetite
- Headache
be life threatening
- A feeling of extreme
- Blurred vision
with shortness of
tiredness or weakness
- Hives, skin rash
breath, low blood
- Fewer white blood cells,
- Wheezing
pressure and a
red blood cells and platelets in the blood.
○ A low number of red blood cells can make you feel tired and weak.
- A low number of white blood cells can make it easier to get infections.
- A low number of platelets causes you to bruise and bleed
- Absence or decrease in the number of sperm which may be temporary or permanent which may decrease the ability to have children
- Absence or decrease in monthly periods and could affect your ability to become pregnant
With High Doses used before marrow transplants:
- Inflammation and/or sores in the mouth, throat and/or esophagus (the passage between the throat and stomach)
- Sudden high fever
- Pain in the abdomen
- Difficulty emptying the bladder
- Feeling the urgency to urinate or pain on urination
- Hair loss
- Inflammation and reddening of the eye
- Inflammation of the skin where the drug comes into contact with the skin
With High Doses used before marrow transplants:
- Inappropriate behavior
- Confusion
- Drowsiness
- Elevation in the blood of certain enzymes and/or bilirubin found in the liver
- Bronze discoloration or darkening of the skin
rapid heart rate
- Swelling and tightening of the throat which can cause difficulty with breathing
- A new cancer or leukemia resulting from this treatment
Risks and side effects related to cyclophosphamide include those which are:
Likely
Less Likely
Rare But Serious
- Loss of appetite
- Nausea
- Vomiting
- Fewer white blood cells in the blood.
- A low number of white blood cells may make it easier to get infections.
- Hair loss
- Decreased ability of the body to fight infection
- Absence or decrease in the number of sperm which may be temporary or permanent which may decrease the ability to have children
- Abnormal hormone function which may lower the level of salt in the blood
- Abdominal pain
- Diarrhea
- Fewer red blood cells and platelets in the blood
- A low number of red blood cells may make you feel tired and weak.
- A low number of platelets may cause you to bruise and bleed more easily.
- Bleeding and inflammation of the urinary bladder
- Absence or decrease monthly periods which may be temporary or permanent and which may decrease the ability to have children
- Temporary blurred vision
- Nasal stuffiness with IV infusions
- Skin rash
- Darkening of areas of the skin and finger nails
- Slow healing of wounds
- Infections
- Heart muscle damage which may occur with very high doses and which may be fatal
- Abnormal heart rhythms
- Damage and scarring of lung tissue which may make you short of breath
- A new cancer or leukemia resulting from this treatment.
- Damage or scarring of urinary bladder tissue
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate chills and fever
- Infertility which is the inability to have children
ARE THERE BENEFITS TO TAKING PART IN THE STUDY?
This experimental treatment may have benefits. This experimental treatment may turn out to be better at treating neuroblastoma than treatments we have used in the past. Unfortunately, there is no guarantee. We may find out that this treatment is not better.
It is hoped that the information learned from this study may help future patients with high-risk neuroblastoma.
WHAT OTHER OPTIONS ARE THERE?
Instead of being in this study, you have these options:
Current standard therapy even if you do not take part in the study.
Other stem cell transplant therapies.
More treatment with standard doses of chemotherapy
No further chemotherapy but just radiation therapy and cis-retinoinc acid therapy.
No further therapy.
You are encouraged to discuss these options with your regular doctor as well as other trusted personal and family advisors.
WHAT ABOUT CONFIDENTIALITY?
Efforts will be made to keep your personal information confidential. We cannot guarantee absolute confidentiality. Your personal information may be disclosed if required by law.
The Children's Oncology Group has received a Certificate of Confidentiality from the federal government, which will help us protect the privacy of our research subjects. Information about the certificate is attached at end of this consent.
Organizations that may inspect and/or copy your research records for quality assurance and data analysis include groups such as:
The Children's Oncology Group
Representatives of the National Cancer Institute (NCI), Food and Drug Administration (FDA), and other U.S. and international governmental regulatory agencies involved in overseeing research
The Institutional Review Board (IRB) of this hospital
Pediatric Central Institutional Review Board (CIRB) of the National Cancer Institute
WHAT ARE THE COSTS?
Taking part in this study may lead to added costs to you or your insurance company. Please ask about any expected added costs or insurance problems. Staff will be able to assist you with this. You or your insurance company will not be charged for research studies performed on blood or bone marrow.
In the case of injury or illness resulting from this study, emergency medical treatment is available but will be provided at the usual charge. No funds have been set aside to pay you in the event of injury. However by signing this form, you are not giving up any legal rights to seek to payment for injury.
You or your insurance company will be charged for continuing medical care and/or hospitalization.
For more information on clinical trials and insurance coverage, you can visit the National Cancer Institute's Web site at http://cancer.gov/clinicaltrials/understanding/insurance-coverage. You can print a copy of the "Clinical Trials and Insurance Coverage" information from this Web site.
There are no plans to pay you for taking part in this study. If this study includes providing specimens to the researcher, there are no plans for you to profit from any new products developed from research done on your specimens
WHAT ARE MY RIGHTS AS A PARTICIPANT?
Taking part in this study is voluntary. You may leave the study at any time. Refusing to take part in the study, or leaving the study will not result in any penalty or loss of benefits to which you are entitled. Your doctor will still take care of you.
We will tell you about new information that may affect your health, welfare, or willingness to stay in this study. A committee outside of COG closely monitors study reports and notifies institutions if changes must be made to the study. Members of COG meet twice a year to evaluate results of treatment and to plan new treatments.
During your follow-up visits after treatment, you may ask to be given a summary of the study results after they are written up. This may be several years after treatment for all people on the study is completed.
WHOM DO I CALL IF I HAVE QUESTIONS OR PROBLEMS?
For questions about the study or a research-related problem, or if you think you have been injured, you may contact Dr. XXXX or your doctor at XXXXX.
If you have any questions about your rights as a research participant or any problems that you feel you cannot discuss with the investigators, you may call XXXX IRB Administrator at (XXXX
If you have any questions or concerns that you feel you would like to discuss with someone who is not on the research team, you may also call the Patient Advocate at XXXX
WHERE CAN I GET MORE INFORMATION?
The COG Family Handbook for Children with Cancer has information about specific cancers, tests, treatment side effects and their management, adjusting to cancer, and resources.
If you are in the United States, you may call the NCI's Cancer Information Service at:
1-800-4-CANCER (1-800-422-6237) or TTY: 1-800-332-8615
A description of this clinical trial will be available at: http://www.ClinicalTrials.gov, as required by U.S. Law. This web site will not include information that can identify you. At most, the web site will include a summary of the results. You can search this web site at any time.
You will get a copy of this form. You may also ask for a copy of the protocol (full study plan).
SIGNATURE
I have been given a copy of all [insert total number of pages] pages of this form. The form includes three (3) attachments.
I agree to take part in this study.
Participant Date
Parent/Guardian Date
Parent/Guardian Date
Physician/PNP obtaining consent Date IRB#
IRB Approved:
Attachment #1
Risks of Chemotherapy Drugs Used
Risks and side effects related to carboplatin include those which are:
Likely
Less Likely
Rare But Serious
- Nausea and vomiting
- fewer red blood cells and white blood cells and platelets in the blood
- a low number of red blood cells can make you feel tired and weak
- a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and bleed more easily
- Abnormal levels of certain salts in the body like sodium and potassium
- Allergic reactions (can be severe and life-threatening causing difficulty in breathing and or a drop in blood pressure)
- Rash
- Metallic taste
- Numbness and tingling in the fingers and toes
- Hair loss
- Constipation or diarrhea
- Pain in your abdomen
- Temporary changes in vision
- Damage to the ear causing hearing and balance problems
- A feeling of weakness and/or tiredness
- Inflammation and/or sores in the mouth (and/or throat and /or esophagus, the tube that leads from the mouth to the stomach) that may make swallowing difficult and are painful (painful mouth sores)
- Damage to the liver
- Damage to the kidney
- Leukemia later in life
Risks and side effects related to etoposide include those which are:
Likely
Less Likely
Rare But Serious
- Nausea and vomiting
- Hair Loss
- A feeling of weakness or tiredness
- fewer red and white blood cells and platelets in the blood
- a low number of red blood cells can make you feel tired and weak
- a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and bleed more easily
- Loss of appetite
- Decreased blood pressure during the infusion which may require treatment
- rashes
- Diarrhea
- Pain in the abdomen
- Mouth sores
- Tingling sensation or loss of sensation in fingers or toes
- A feeling of extreme tiredness or weakness
- The finger or toe nails may loosen from their nail beds
- Inflammation of the vein through which the medication was given
- Chest pain
- Damage to the liver
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, chills and fever
- A new cancer or leukemia resulting from this treatment
- Severe rashes which can result in loss of skin and damage to mucous membranes
- Absence or decrease monthly periods which may be temporary or permanent and which may decrease the ability to have children
- Damage to the heart muscle which may make you feel tired, weak, feel short of breath, and retain fluid
Risks and side effects related to isotretinoin (Accutane) include those which are:
Likely
Less Likely
Rare But Serious
- Dryness of your skin and mucous membranes
- Dry, cracked and bleeding lips
- An increased tendency to sun burn
- Bloody nose from dry membranes of the nose
- Aches and pains in the joints
- Back pain
- Elevation of the fats in your blood
- Increase in calcium in your blood which may require decreasing the dose
- Rash and itching
- Headache
- Increase in cholesterol and a decrease in the good fat in the blood
- Red eyes
- Elevation in the blood of certain enzymes found in the liver which may mean liver irritation or damage
- Fewer red blood cells and white blood cells and platelets in the blood ○ a low number of red blood cells can make you feel tired and weak
○ a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, chills and fever
- Irritation of the small airways in your lungs that can make you cough and wheeze
- An allergic reaction in the blood vessels of the skin which turn the skin red, inflamed and bumpy and which may lead to skin breakdown
- A severe lowering of the white blood count which can make you very susceptible to infections
- An increase in a laboratory test on your blood that may measure some non specified inflammation which may or may not be of any importance
bleed more easily
- Too many platelets in the blood
- Loss or thinning of hair
- Appetite disturbances causing you not to feel hungry or to feel unusually hungry
- Weight loss
- Increase in blood sugar levels
- A darkening or lightening of your skin
- Finger and toe nail changes including breaking or splitting more easily
- The sudden appearance of little yellow raised bumps on the skin usually because the cholesterol in the blood is too high (xanthomas)
- Dizziness
- Difficulty falling asleep or staying asleep and strange dreams
- A feeling of tiredness or not feeling well
- Nervousness
- Numbness and tingling in the fingers and toes
- Difficulty hearing clearly or a ringing in the ears
- Changes in vision including more difficulty seeing at night, blurred vision, changes in color vision, pain or squinting in bright light, and cataract formation
- Fluid retention
- Chest pain
- Inflammation of the gums
- A dry throat which could lead to a change in your voice and more throat infections
- Slowed growth
- Irregular periods
which could be life threatening
- Convulsions
- Brain swelling that can give you symptoms of severe headache, nausea and vomiting, and changes to your vision including blurriness and pressure behind the eyes
- Life threatening or fatal changes in moods have occurred including severe depression or feelings of suicide and feelings of aggressiveness and violent behavior
- Thinning of the bone (osteoporosis) which could lead to weakness of the bone, bone fractures or delay in healing of fractures
- Inflammation of the pancreas which can lead to severe abdominal pain and in some very rare cases can be fatal
- Damage to the muscle which can release a protein that can cause severe damage to the kidneys
- Inflammation of the intestinal tract which can result in diarrhea and bleeding
- This drug can cause severe birth defects in a developing fetus, if you are capable of becoming pregnant or of child bearing age, you must practice 2 forms of reliable birth control, sign the Patient Information/Informed Consent form(s), have
- Mild kidney damage which could lead to blood or protein in the urine or renal stones
- Extra bone growth along the spine and a tendency for calcium deposits in the tendons and ligaments where they attach to the bone which can lead to pain or stiffness and arthritis of the back and tendonitis
regular pregnancy tests, be able to keep appointments and agree to follow the iPLEDGE program steps (a special program required by the manufacturers and approved by the Food and Drug Administration (FDA) which your doctor will explain to you).
Risks and side effects related to melphalan include those which are:
Likely
Less Likely
Rare But serious
- Loss of Appetite
- Nausea and/or vomiting
- Low levels of salt in the blood which may need to be treated (usually associated with high doses)
- Diarrhea
- Inflammation and/or sores in the mouth (and/or throat and/or esophagus, the tube that leads from the mouth to the stomach) that may make swallowing difficult and are painful (painful mouth sores)
- Temporary hair loss
- Fewer white blood cells, red blood cells and platelets in the blood
- a low number of white blood cells can make it easier to get infections
- a low number of red blood cells can make you feel tired and weak
- a low number of platelets causes you to bruise and bleed more easily
- Absence of menstrual cycles (periods) and damage to the ovaries that may decrease the ability to have children in the future
- Absence or decrease in the number of sperm which may be temporary or permanent which may decrease the ability to have children
- Inability to have children (infertility)
- Sweating
- Itching
- Low blood pressure
- Abnormal heart rate (usually associated with high doses)
- Damage to the skin if the medication leaks from the vein
- Increase in the blood of certain enzymes or bilirubin (a substance that comes from the liver breaking down waste products) which could indicate liver irritation or damage
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, chills and fever
- Seizures
- Damage to the liver which can lead to inflammation and/or scarring which could lead to a yellow appearing skin, and fluid collection in the abdomen (belly) which makes it look larger
- Severe damage to the bone marrow which could lead to low numbers of white blood cells, red blood cells and platelets and could be permanent
- Sudden damage to the red blood cells (hemolytic anemia) which could cause a rapid decrease in the number of red blood cells such that you would be tired and weak and feel short of breath and may require a blood transfusion
- Inflammation and/or scarring of the lungs that can lead to fluid in the lungs and affect your ability to breath and the levels of oxygen in your blood making you short of breath
- A new cancer or leukemia resulting from this treatment
Risks and side effects related to myeloid growth factors (filgrastim (G-CSF) or pegfilgrastim) include those which are:
Likely
Less Likely
Rare but serious
- Aching or pain in the bones
- Pain, redness, itching, and hardening of the skin and bruising at the site of the injection
- Headache
- Higher than normal levels of liver enzymes in the blood which may indicate liver irritation or damage
- Increase of uric acid in the blood
- A low number of platelets in the blood which may cause you to bruise and bleed more easily
- Low fever
- Enlargement of the spleen (an organ in the abdomen/belly which stores blood cells) which may cause pain in the abdomen or left shoulder
- Higher than normal white blood count
- Skin condition marked by fever and painful skin lesions that appear mainly on the face, neck, back and arms
- Rash or worsening of rash
- Inflammation of blood vessels in the skin leading to a raised purple rash and bruising has been seen mainly in patient who are treated for a long time
- Overall reddening with feelings of warmth
- Allergic reactions which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, hives, itching, and facial swelling.
- Serious allergic reaction which can be life threatening with rapid build-up of fluid under the skin, in the lining of the intestine, and possibly in the throat or swelling of the tongue which could make it difficult to breath
- If you are known to have sickle cell disease, filgrastim or pegfilgrastim may cause a sickle cell crisis.
- Severe damage to the spleen (an organ in the abdomen/belly which stores blood cells) which could lead to pain and loss of blood into the abdomen (belly) and maybe life threatening
- Difficulty breathing and lung damage that may be due to the white blood cells that are stimulated by filgrastim or pegfilgrastim traveling to the lungs when they are inflamed or infected.
- A blood disorder or leukemia that has only been seen in patients with certain immune disorders who are treated for a very long time
Children's Oncology Group
Risks and side effects related to mesna include those which are:
Likely
Less Likely
Rare But Serious
- Nausea.
- Vomiting
- Stomach pain.
- Headache.
- Pain in arms, legs and joints.
- Tired feeling.
- Rash.
- Temporary low blood pressure.
- Diarrhea.
- Fever
- Facial flushing with red cheeks
- Nervousness
- Dizziness
- Confusion
- Swelling around the eyes
- Coughing
- Rapid heart rate
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate chills and fever
Attachment #2
Risks of Radiation Therapy and Peripheral Blood Stem Cell Harvesting and Reinfusion
Radiation Therapy Risks
The risks of radiation therapy depend on the parts of the body being treated. Some possible risks are described below, but you should talk to your child's doctor to see which apply to your child. Radiation therapy can cause nausea, vomiting, diarrhea, red or dry skin, low blood counts, hair loss (permanent or temporary), jaw pain and swelling, temporary weakness or loss of sensation. Some patients have a week or two of low grade fever and sleepiness can occur six to eight weeks after radiation therapy is done. Damage to body organs such as the brain, eyes, heart, lung, liver, and kidneys can occur. Radiation therapy can also cause abnormal bone growth. There is also a small chance that radiation can cause another type of tumor years later.
Peripheral Blood Stem Cell Harvesting and Reinfusion
These procedures are usually safe. Side effects that can occur during PBSC collection include nausea, vomiting, fainting or dizziness, seizures, skin rash, hives, flushing (redness and warmness of the skin, usually the face), blood loss, and infection. Tingling of the lips, muscle cramping and, very rarely, changes in the heart rhythm can occur. These can be prevented or made milder by giving calcium supplements, either by mouth or IV. Very rarely, (less than 1 in 1,000 procedures), clotting may occur in the apheresis machine or in a patient and is potentially life-threatening. To reduce the risk of clotting, you will be given a drug called ACD (acid-citratedextrose). This drug may increase the risk of bleeding and may cause temporary tingling of the lips and limbs, muscle cramping, seizures, or changes in the heart rhythm.
The risks associated with infusing the cells back into your body include dark urine, nausea, vomiting, fever, chills, and high blood pressure. All of these are temporary and go away after the infusion is done. As with any procedure, there may be side effects we do not expect.
Attachment #3
Certificate of Confidentiality Information
The Children's Oncology Group has received a Certificate of Confidentiality from the federal government, which will help us protect the privacy of our research subjects. The Certificate protects against the involuntary release of information about subjects collected during the course of our covered studies. The researchers involved in the studies cannot be forced to disclose the identity or any information collected in the study in any legal proceedings at the federal, state, or local level, regardless of whether they are criminal, administrative, or legislative proceedings. However, the subject or the researcher may choose to voluntarily disclose the protected information under certain circumstances. For example, if the subject or his/her guardian requests the release of information in writing, the Certificate does not protect against that voluntary disclosure. Furthermore, federal agencies may review our records under limited circumstances, such as a DHHS request for information for an audit or program evaluation or an FDA request under the Food, Drug and Cosmetics Act. The Certificate of Confidentiality will not protect against the required reporting by hospital staff of information on suspected child abuse, reportable communicable diseases, and/or possible threat of harm to self or others.
SAMPLE INFORMED CONSENT / PARENTAL PERMISSION FOR PARTICIPATION IN RESEARCH
This model informed consent form has been reviewed by the DCT/NCI and is the official consent document for this study. Local IRB changes to this document are allowed. (Institutions should attempt to use sections of this document which are in bold type in their entirety.) Editorial changes to these sections may be made as long as they do not change information or intent. If the local IRB insists on making deletions or more substantive modifications to the risks or alternatives sections, they must be justified in writing by the investigator and approved by the IRB .
ANBL0532, Phase III Randomized Trial of Single vs. Tandem Myeloablative Consolidation Therapy for High-Risk Neuroblastoma:
PART 2 B: CONSENT FOR PATIENTS TO BE NON-RANDOMLY ASSIGNED TO SINGLE MYELOABLATIVE CONSOLIDATION THERAPY
Consolidation and Maintenance Phases of Therapy for High Risk Neuroblastoma (Please note that this consent MUST be signed before the patient proceeds for consolidation therapy)
When the word "you" appears in this consent form, it refers to you or your son or daughter; "we" means the doctors and other staff.
WHY ARE YOU BEING INVITED TO TAKE PART IN THIS STUDY?
This is a clinical trial, a type of research study. Your study doctor will explain the clinical trial to you.
The Children's Oncology Group (COG) is carrying out this study. COG is an international research group that conducts clinical trials for children with cancer at more than 200 hospitals in the United States, Canada, Australia, New Zealand, the Netherlands, and Switzerland. It is common medical practice to treat children with cancer on research studies like this one.
You are being asked to take part in this study because you have high risk neuroblastoma and you have participated in Part 1 of this study. Neuroblastoma is a type of cancer. You have the type of neuroblastoma that is called High Risk because your tumor has spread from where it started from or because your type of tumor is harder to treat.
It is common to enroll children and adolescents with cancer in a clinical trial that seeks to improve cancer treatment over time. Clinical trials include only people who choose to take part. You have a choice between a standard treatment for High Risk neuroblastoma and this clinical trial. Please take your time to make your decision about taking part in this clinical trial. You may discuss your decision with your friends and family. You can also discuss it with your health care team. We encourage you to include your child in the discussion and decision to the extent that she or he is able to understand and take part. If you have any questions, you can ask your study doctor for more explanation.
WHAT IS THE CURRENT STANDARD OF TREATMENT FOR THIS DISEASE?
The treatment for neuroblastoma includes 3 parts (phases) of therapy called Induction, Consolidation and Maintenance. During Induction therapy anti-cancer drugs (chemotherapy) and surgery are used to kill and remove as much tumor as possible. Blood stem cells are collected during the Induction phase of therapy. After collection the blood stem cells are frozen
and stored to be used during Consolidation phase of treatment. Blood stem cells are the cells that create new blood cells, such as red blood cells, white blood cells, and platelets. Blood stem cells can be collected from the blood by using a machine that can separate out the part of blood that has stem cells and then return the remaining blood back to the patient. You have already received this part of therapy.
During the Consolidation phase of treatment extremely high doses of chemotherapy are given to better kill any remaining neuroblastoma cells. It is standard therapy to give one cycle of this extremely high dose chemotherapy. The high doses of chemotherapy destroy healthy bone marrow. Bone marrow is the soft tissue in the hollow of flat bones of the body that produces new blood cells. The peripheral blood stem cells that were stored during the Induction phase of treatment are given back to the patient after the high dose Consolidation chemotherapy. These stems cells allow the bone marrow to return to normal so that new blood cells can be made. This type of therapy is called a Hematopoietic Stem Cell Transplant. Once the patient has healed from the effects of high doses of chemotherapy, radiation therapy is given to the first place the tumor was found and to any additional places where the tumor was found after Induction therapy.
During the third phase of therapy, Maintenance, an oral drug, called cis-retinoic acid (Accutane) is given.
WHY IS THIS STUDY BEING DONE?
Most of the patients enrolled on this study have very high risk neuroblastoma. The main goal of this study applies to them. That goal is to find out if using 2 cycles of high dose Consolidation chemotherapy, instead of 1 cycle, will decrease the chance that the neuroblastoma will grow back after therapy. However, some types of high risk neuroblastoma have a lower risk of having tumor grow back.
These types of neuroblastoma are described as being in one of two groups. These groups are:
Patients who are between the ages of 12 and 18 months at diagnosis of neuroblastoma. Their neuroblastoma tumor has spread to distant sites (stage 4), does not have increased levels of the MYCN gene, but does have other high risk biologic characteristics.
Patients who are greater than 18 months of age at diagnosis of neuroblastoma. Their neuroblastoma tumor has spread locally (stage 3), does not have increased levels of the MYCN gene but does have other high risk biologic characteristics.
Very few patients with high risk neuroblastoma (about ) have these types of neuroblastoma. Previous clinical trials have not always used high dose Consolidation chemotherapy to treat these types of high risk neuroblastoma. We need to learn more about the best way to treat this smaller group. We want to evaluate how patients in this group respond to treatment after 1 cycle of high dose Consolidation therapy.
This research study will also find out if patients whose tumor did not disappear with initial induction therapy will need a higher dose of radiation therapy.
The treatment is described in two separate consent forms. You have already read the first consent form (Part 1) and completed Part 1 of treatment. We are now presenting Part 2 (the second consent form) of the study. The second consent (Part 2) will discuss the Consolidation and Maintenance phases of therapy.
During this part of the study, we will:
Find out if patients with high-risk neuroblastoma who have a lower risk of the tumor coming back can be treated with 1 cycle of high dose Consolidation chemotherapy.
Study whether a higher dose of radiation, given only to patients with tumor remaining after induction therapy, will decrease the chance that tumor will grow back.
Study whether your immune system (blood cells that help fight infection) can produce cells that can recognize and kill neuroblastoma.
Study whether new tests can be used to find small amounts of neuroblastoma cells.
Study how cis-retinoic acid (Accutane) is broken down by your body and study whether your genes (genes direct activities of cells) effect how you respond to cisretinoic acid therapy.
HOW MANY PEOPLE WILL TAKE PART IN THIS STUDY?
About 664 subjects will take part in this study. The study will last about years.
WHAT WILL HAPPEN ON THIS STUDY?
If you participate in this part of the study you will be given the following treatment plan.
Treatment Plan
The treatment plan involves cancer fighting medicine called chemotherapy plus radiation therapy. The treatment on this part of the clinical trial takes about 9 months.
Myeloablative therapy means that you will receive very high doses of chemotherapy that will destroy normal bone marrow cells.
You will receive consolidation treatment after you have finished induction therapy and signed this consent.
Diagram of treatment
A diagram of part 2 of the treatment can be seen below.
Children's Oncology Group
Treatment Plan Tables
The following drug therapies will be used for Single Myeloablative Consolidation.
Various methods will be used to give drugs:
PO - Drug is given by tablet or liquid swallowed through the mouth (PO).
IV - Drug is given using a needle or tubing inserted into a vein. It can be given by IV push over several minutes or by infusion over minutes or hours
SUBQ - Drug is given by injecting a needle into the tissue just under the skin (SUBQ shot)
Most drugs on this study will be given using a needle or tubing inserted into a vein (IV).
Treatment
After you finish induction therapy, you will get one cycle of very high dose chemotherapy and a hematopoietic stem cell transplant. The transplant will begin with treatment with very high doses of the chemotherapy drugs carboplatin, etoposide and melphalan. Etoposide and carboplatin are given as a continuous infusion for 4 consecutive days. The drug melphalan is given as a daily dose for 3 days. After the high dose chemotherapy treatment, your stored stem cells will be given IV (through the central line). These stem cells were collected from you during the induction phase of therapy and frozen. This is called a Hematopoietic Stem Cell Transplant. You will be given filgrastim (GCSF) starting on the same day you receive your stem cells and continuing until your white blood cells have returned to a safe number, usually about 2 weeks.
Drug
How the drug will be given
Days
Carboplatin
IV over 24 hours
Etoposide
IV over 24 hours
Melphalan
IV over 15-30 minutes
Blood Stem Cells
IV
0
Filgrastim (GCSF)
SUBQ (or) IV
Radiation Therapy
It is standard to give radiation treatment to the area of the main tumor and to areas that still showed signs of active disease at the end of Induction phase of treatment. Radiation therapy will begin after you have gotten better from the immediate side effects of the high doses of chemotherapy. Radiation therapy usually starts about a month after the transplant. The radiation therapy doctors will discuss this with you in more detail. The radiation is given in one short session each day, for a period of up to 3 weeks. Radiation to the stomach may cause nausea and vomiting, and often causes the blood counts to drop temporarily, which may require treatment with filgrastim (GCSF) or transfusions (give you blood). Other risks are discussed below.
You will get a higher dose of radiation treatment if x -ray tests performed at the end of Induction therapy still show tumor (a lump). The higher dose of radiation will only be given to the area where tumor is present. This higher dose of radiation is experimental. We are studying whether this higher dose of radiation will decrease the risk of tumor growing back.
MAINTENANCE PHASE OF THERAPY
Maintenance therapy will begin after you have recovered from effects of radiation therapy. The use of an immune therapy that targets neuroblastoma (a medicine called chimeric 14.18 antibody) combined with the prior standard therapy of cis-Retinoic acid (Accutane) has recently been shown to improve survival for children with high risk neuroblastoma. Chimeric 14.18 antibody is only available to patients with high risk neuroblastoma by enrolling onto another clinical trial. It is strongly encouraged that you speak with your physician about receiving chimeric 14.18 therapy. If you are not able to get ch14.18 therapy or choose not to get it, then you will get cis-Retinoic acid (Accutane) therapy alone. Cis-Retinoic acid is given by mouth, twice daily for 2 weeks, followed by two weeks without the drug. This cycle will be repeated 6 times for a total of 6 months of treatment.
Standard Medical Tests
Before treatment on this study begins, and while receiving treatment, you will have a series of standard medical tests:
Physical exam
Blood Tests
Bone marrow tests
Tests of vision and hearing
Urine tests
Various scans (x-ray tests to see where the tumor is in your body)
Tests of kidney function
Tests of lung and heart function
Hearing tests
Dental exams
Research study tests and procedures
The following tests will be done because you are part of this study. These tests are not part of standard care.
Some copies of the scans (x-rays) used to make the diagnosis of your disease and to see how your tumor responded to therapy will be sent to a central review center as part of COG quality control.
We would like to collect additional blood to see if certain genes (genes direct the activities of cells) will effect whether you experience bad effects after chemotherapy, to see if your immune system is able to recognize neuroblastoma tumor cells and to test new methods for finding tumor cells. We will get all blood specimens through your central venous catheter. We will try to get these additional blood samples when you are already having blood drawn for routine purposes.
We would like to use any leftover portions of your bone marrow that is not needed to treat you to carry out special biology research studies to learn more about neuroblastoma. These studies will test new methods for finding tumor cells.
Blood tests
Before starting cis-retinoic acid (Accutane) treatment, we would like to take additional blood (20 teaspoons) to see if your immune cells can recognize neuroblastoma tumors.
After you have received the first 14 days of cis-retinoic acid, we would like to take additional blood ( ) to look at how certain genes (genes direct the activities of cells) may affect whether you have side effects.
At the end of Consolidation phase of therapy (after completing radiation therapy) and at the end of Maintenance therapy, we would like to obtain additional blood ( teaspoon) to test new methods of finding neuroblastoma.
Bone marrow tests
We would also like to take 0.5 ml (1/10 tsp) of extra bone marrow at the end of Consolidation therapy and at the end of Maintenance therapy to study new methods of finding tumor cells. This bone marrow will be taken when we are normally taking a sample to see how you are responding to therapy. The extra bone marrow will be shipped to a central lab for storage.
You and your physicians will not be given the results of these blood and bone marrow tests. You will not benefit from allowing us to perform these extra studies. We will try to take the blood samples when we are already taking blood for standard therapy. The blood will be taken from your central line and will not require an extra blood draw. You can ask that we stop collecting these specimens at any time during your treatment. You will continue to get treatment on the study whether or not you allow us to collect these additional samples.
Please read each sentence below and think about your choice. After reading each sentence, choose "Yes" or "No" then add your initials and date after your answer. No matter what you decide to do, it will not affect your care. If you have any questions, please talk to your doctor or nurse, or call our research review board at IRB's phone number included in this consent.
I agree to the have additional bone marrow obtained for research purposes (to find new ways to detect neuroblastoma).
Yes
No Initials
I agree to have additional blood obtained for research purposes (to test for immune cells that recognize neuroblastoma).
Yes
No Initials
I agree to have additional blood obtained for research purposes (to find new ways to detect neuroblastoma).
Yes
No Initials
I agree to have additional blood obtained for research purposes (to test how cis-RA is broken down).
Yes
No Initials
HOW LONG WILL I BE ON THIS STUDY?
The Consolidation phase of therapy takes about 2-3 months. The Maintenance phase of therapy will last an additional 6 months.
We will continue to collect some medical information about how you are doing for 10 years after you enter the study. Keeping in touch with you and checking on how your health is every year for a while after you complete treatment helps us understand the long-term effects of the study.
Your doctor or the study doctor may decide to take you off this study for the following reasons:
he/she believes that it is your best interest
your disease comes back during treatment
you experiences side effects from the treatment that are considered too severe
new information becomes available that shows that another treatment would be better for you
You can stop participating at any time. However, if you decide to stop participating in the study, we encourage you to talk to the study doctor and your regular doctor first.
WHAT ARE THE RISKS OF THE STUDY?
Treatment Risks
All people who have cancer treatment are at risk of having side effects. In addition to killing tumor cells, cancer chemotherapy can damage normal tissue and produce side effects. Side effects are usually reversible when the medication is stopped but occasionally persist and cause serious complications. A person can die from these and other complications.
Common side effects include nausea, vomiting, hair loss, and fatigue. Drugs may be given to prevent or decrease nausea and vomiting. Hair loss is usually temporary but on very rare occasions it may be permanent. Some chemotherapy may lead to sterility. Sterility means that you will not be able to have children. There is also the possibility that a second cancer may develop years later as a result of the chemotherapy. The risks of the individual drugs given as standard treatment are listed on the tables in Attachment #1. Side effects can be increased when chemotherapy drugs are combined.
The most common serious side effect from cancer treatment is lowering of the number of blood cells resulting in anemia, increased chance of infection, and bleeding tendency. Low blood counts are described in your Family Handbook for Children with Cancer. The high doses of chemotherapy used during the Consolidation phase of treatment will cause very severe lowering of the blood cell numbers. You will be at risk to develop a life-threatening infection and bleeding following the very high doses of chemotherapy. To limit the risk of infection, your child will remain in a special hospital room until blood counts return to safe levels. Your stem cells that were previously collected and stored will be used to help your body make new blood cells.
It is possible that it will take a very long time for your blood cells to be made. This can happen if the stem cells were injured in your body even before they were removed or if the stem cells are injured during the collection, freezing or thawing processes. If this happens, you will have a high chance that you will develop a severe infection and/or bleeding. This is very unlikely to happen, but would almost certainly be fatal. Even after your body makes blood cells after the transplant, your ability to fight infection will be low for weeks to months after the transplant is complete. You will be more likely to develop an infection in the months after transplant, especially from viruses.
There is also a small risk of severe organ damage, especially to liver and kidney, with an even smaller risk to the heart and lungs. Though rare, the kidney damage can be severe enough to require dialysis. The liver damage can also be severe, resulting in jaundice and, occasionally, complete failure of the liver to function. This damage may be reversible or irreversible, and if irreversible, the liver failure would be fatal.
There is a chance that the stem cells may contain tumor cells. These cells could result in the tumor coming back.
These problems alone or in combination may be severe enough to be life-threatening. The purpose of using stem cells is to decrease the chance that these problems will happen. Your doctor will also give you antibiotic and blood transfusions to decrease risk for infection or bleeding.
Months or years after treatment other side effects may appear.
We know that the chemotherapy drugs cyclophosphamide, melphalan and etoposide can cause leukemia.
Radiation therapy may cause bone cancers or other kinds of sarcomas.
Cyclophosphamide and melphalan will cause damage to the sexual glands. As a result, if you are a boy, you will almost certainly not be able to have biologically related children, and, if you are girl, you are likely to need hormones or other interventions to have children.
Reproductive risks:
It is unknown what effect(s) these treatments may have on an unborn child. As the drugs and/or radiation therapy in this study can affect a developing fetus (unborn baby in the womb), you should not become pregnant or father a baby while on this study. For this reason, if you are of child-bearing age, you will be asked to practice an effective method of birth control while participating on this study. The use of Isotretinoin can cause birth defects to unborn children if taken during pregnancy.
Ask about counseling and more information about preventing pregnancy. Having the chemotherapy described may cause infertility (being less able to produce a viable egg or sperm) or sterility (being unable to produce a viable egg or sperm). We will talk to males who have reached puberty about sperm banking.
For Women:
The treatment on this study can affect an unborn child. You should not become pregnant or breast feed your baby while being treated on this study. If you are sexually active and are at risk of getting pregnant, you and your male partner(s) must use an effective method to avoid pregnancy or you must not have sex. The study doctor will talk to you about acceptable methods to avoid pregnancy while you are being treated on this study. You will have to use the chosen method to avoid pregnancy or abstain (not have sexual intercourse) the whole time you are being treated on this study. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid pregnancy or abstain. Natural family planning and the rhythm method will not be permissible means of avoiding pregnancy during study participation. If you have questions about this or want to change your method to avoid pregnancy during therapy, please ask your doctor. If you become pregnant during the research study, please tell the investigator and your doctor immediately.
If you are nursing a baby, the drugs used in this research could pass into the breast milk. You should not nurse your baby for the whole time you are getting the study medicines. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid nursing.
For Men:
The treatment on this study can damage sperm. You should not father a child while on this study as the treatment may indirectly affect an unborn child. If you are sexually active and are at risk of causing a pregnancy, you and your female partner(s) must use a method to avoid pregnancy that works well or you must not have sex. The investigator will talk to you about the acceptable methods to avoid pregnancy while you are being treated on this study. You will have to use the chosen method to avoid pregnancy or abstain (not have sexual intercourse) the whole time you are being treated on this study. You may need to continue this for a while, even after you finish the cancer treatment, so talk to your doctor about the length of time you need to avoid pregnancy or abstain.
Natural family planning and the rhythm method will not be permissible means of avoiding pregnancy during study participation. If you have questions about this or want to change your method to avoid pregnancy during therapy, please ask your doctor. If your partner becomes pregnant during the research study, please tell the investigator and your doctor immediately.
There is a risk that the treatment plan will not cure the cancer or that the cancer can go away after the treatment and then come back at a later date.
Risks of Study
The side effects of high dose chemotherapy are more severe than the side effects of standard doses of chemotherapy.
You may get a higher dose of radiation if you still had tumor remaining after finishing the Induction phase of treatment. For most patients, the area receiving the additional radiation dose will be small. However, it is possible that the use of higher doses of radiation may be more likely to cause the side effects of radiation that are described in Attachment #2.
Please see the attachment at the end of the consent (Attachment #2) for more information regarding the risks associated with radiation therapy and Peripheral Blood Stem Cell Harvesting and Reinfusion.
Information may also be found in the COG Family Handbook
For more information about risks and side effects, ask your child's study doctor.
In addition to the risks described above, there may be unknown risks, or risks that we did not anticipate, associated with being in this study.
Your physician will be checking closely to see if any of these side effects are occurring. Routine physical exams and laboratory tests will be done to monitor the effects of treatment. Side effects usually disappear after the treatment is completed.
Treatment on this study may have risks we don't know about. We may stop this treatment if we learn of serious, unexpected risks. We will explain the effects of stopping, and we will offer other treatments.
ARE THERE BENEFITS TO TAKING PART IN THE STUDY?
This experimental treatment may have benefits. This experimental treatment may turn out to be better at treating neuroblastoma than treatments we have used in the past. Unfortunately, there is no guarantee. We may find out that this treatment is not better.
It is hoped that the information learned from this study may help future patients with high-risk neuroblastoma.
WHAT OTHER OPTIONS ARE THERE?
Instead of being in this study, you have these options:
Current standard therapy even if you do not take part in the study.
Other stem cell transplant therapies.
More treatment with standard doses of chemotherapy
No further chemotherapy but just radiation therapy and cis-retinoinc acid therapy.
No further therapy.
You are encouraged to discuss these options with your regular doctor as well as other trusted personal and family advisors.
WHAT ABOUT CONFIDENTIALITY?
Efforts will be made to keep your personal information confidential. We cannot guarantee absolute confidentiality. Your personal information may be disclosed if required by law.
The Children's Oncology Group has received a Certificate of Confidentiality from the federal government, which will help us protect the privacy of our research subjects. Information about the certificate is attached at end of this consent.
Organizations that may inspect and/or copy your research records for quality assurance and data analysis include groups such as:
The Children's Oncology Group
Representatives of the National Cancer Institute (NCI), Food and Drug Administration (FDA), and other U.S. and international governmental regulatory agencies involved in overseeing research
The Institutional Review Board (IRB) of this hospital
Pediatric Central Institutional Review Board (CIRB) of the National Cancer Institute
WHAT ARE THE COSTS?
Taking part in this study may lead to added costs to you or your insurance company. Please ask about any expected added costs or insurance problems. Staff will be able to assist you with this. You or your insurance company will not be charged for research studies performed on blood or bone marrow.
In the case of injury or illness resulting from this study, emergency medical treatment is available but will be provided at the usual charge. No funds have been set aside to pay you in the event of injury. However by signing this form, you are not giving up any legal rights to seek to payment for injury.
You or your insurance company will be charged for continuing medical care and/or hospitalization.
For more information on clinical trials and insurance coverage, you can visit the National Cancer Institute's Web site at http://cancer.gov/clinicaltrials/understanding/insurance-coverage. You can print a copy of the "Clinical Trials and Insurance Coverage" information from this Web site.
There are no plans to pay you for taking part in this study. If this study includes providing specimens to the researcher, there are no plans for you to profit from any new products developed from research done on your specimens
WHAT ARE MY RIGHTS AS A PARTICIPANT?
Taking part in this study is voluntary. You may leave the study at any time. Refusing to take part in the study, or leaving the study will not result in any penalty or loss of benefits to which you are entitled. Your doctor will still take care of you.
We will tell you about new information that may affect your health, welfare, or willingness to stay in this study. A committee outside of COG closely monitors study reports and notifies institutions if changes must be made to the study. Members of COG meet twice a year to evaluate results of treatment and to plan new treatments.
During your follow-up visits after treatment, you may ask to be given a summary of the study results after they are written up. This may be several years after treatment for all people on the study is completed.
WHOM DO I CALL IF I HAVE QUESTIONS OR PROBLEMS?
For questions about the study or a research-related problem, or if you think you have been injured, you may contact Dr. XXXX or your doctor at XXXXX.
If you have any questions about your rights as a research participant or any problems that you feel you cannot discuss with the investigators, you may call XXXX IRB Administrator at (XXXX
If you have any questions or concerns that you feel you would like to discuss with someone who is not on the research team, you may also call the Patient Advocate at XXXX
WHERE CAN I GET MORE INFORMATION?
The COG Family Handbook for Children with Cancer has information about specific cancers, tests, treatment side effects and their management, adjusting to cancer, and resources.
A description of this clinical trial will be available at: http://www.ClinicalTrials.gov, as required by U.S. Law. This web site will not include information that can identify you. At most, the web site will include a summary of the results. You can search this web site at any time.
You will get a copy of this form. You may also ask for a copy of the protocol (full study plan).
SIGNATURE
I have been given a copy of all [insert total number of pages] pages of this form. The form includes three (3) attachments.
I agree to take part in this study.
Participant Date
Parent/Guardian Date
Parent/Guardian Date
Physician/PNP obtaining consent Date IRB#
IRB Approved:
Attachment #1
Risks of Chemotherapy Drugs Used
Risks and side effects related to carboplatin include those which are:
Likely
Less Likely
Rare But Serious
- Nausea and vomiting
- fewer red blood cells and white blood cells and platelets in the blood
- a low number of red blood cells can make you feel tired and weak
- a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and bleed more easily
- Abnormal levels of certain salts in the body like sodium and potassium
- Allergic reactions (can be severe and life-threatening causing difficulty in breathing and or a drop in blood pressure)
- Rash
- Metallic taste
- Numbness and tingling in the fingers and toes
- Hair loss
- Constipation or diarrhea
- Pain in your abdomen
- Temporary changes in vision
- Damage to the ear causing hearing and balance problems
- A feeling of weakness and/or tiredness
- Inflammation and/or sores in the mouth (and/or throat and /or esophagus, the tube that leads from the mouth to the stomach) that may make swallowing difficult and are painful (painful mouth sores)
- Damage to the liver
- Damage to the kidney
- Leukemia later in life
Risks and side effects related to etoposide include those which are:
Likely
Less Likely
Rare But Serious
- Nausea and vomiting
- Hair Loss
- A feeling of weakness or tiredness
- fewer red and white blood cells and platelets in the blood
- a low number of red blood cells can make you feel tired and weak
- a low number of white blood cells can make it easier to get infections
- a low number of platelets causes you to bruise and bleed more easily
- Loss of appetite
- Decreased blood pressure during the infusion which may require treatment
- rashes
- Diarrhea
- Pain in the abdomen
- Mouth sores
- Tingling sensation or loss of sensation in fingers or toes
- A feeling of extreme tiredness or weakness
- The finger or toe nails may loosen from their nail beds
- Inflammation of the vein through which the medication was given
- Chest pain
- Damage to the liver
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, chills and fever
- A new cancer or leukemia resulting from this treatment
- Severe rashes which can result in loss of skin and damage to mucous membranes
- Absence or decrease monthly periods which may be temporary or permanent and which may decrease the ability to have children
- Damage to the heart muscle which may make you feel tired, weak, feel short of breath, and retain fluid
Risks and side effects related to isotretinoin (Accutane) include those which are:
Likely
Less Likely
Rare But Serious
- Dryness of your skin and mucous membranes
- Dry, cracked and bleeding lips
- An increased tendency to sun burn
- Bloody nose from dry membranes of the nose
- Aches and pains in the joints
- Back pain
- Elevation of the fats in your blood
- Increase in calcium in your blood which may require
- Rash and itching
- Headache
- Increase in cholesterol and a decrease in the good fat in the blood
- Red eyes
- Elevation in the blood of certain enzymes found in the liver which may mean liver irritation or damage
- Fewer red blood cells and white blood cells and platelets in the blood ○ a low number of red blood cells can make you feel tired and weak
○ a low number of white blood cells can make it easier to get infections
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, chills and fever
- Irritation of the small airways in your lungs that can make you cough and wheeze
- An allergic reaction in the blood vessels of the skin which turn the skin red, inflamed and bumpy and which may lead to skin breakdown
- A severe lowering of the white blood count which
decreasing the dose
An increase in a laboratory test on your blood that may measure some non specified inflammation which may or may not be of any importance
○ a low number of platelets causes you to bruise and bleed more easily
Too many platelets in the blood
Loss or thinning of hair
Appetite disturbances causing you not to feel hungry or to feel unusually hungry
Weight loss
Increase in blood sugar levels
A darkening or lightening of your skin
Finger and toe nail changes including breaking or splitting more easily
The sudden appearance of little yellow raised bumps on the skin usually because the cholesterol in the blood is too high (xanthomas)
Dizziness
Difficulty falling asleep or staying asleep and strange dreams
A feeling of tiredness or not feeling well
Nervousness
Numbness and tingling in the fingers and toes
Difficulty hearing clearly or a ringing in the ears
Changes in vision including more difficulty seeing at night, blurred vision, changes in color vision, pain or squinting in bright light, and cataract formation
Fluid retention
Chest pain
Inflammation of the gums
A dry throat which could lead to a change in your voice and more throat infections
can make you very susceptible to infections which could be life threatening
Convulsions
Brain swelling that can give you symptoms of severe headache, nausea and vomiting, and changes to your vision including blurriness and pressure behind the eyes
Life threatening or fatal changes in moods have occurred including severe depression or feelings of suicide and feelings of aggressiveness and violent behavior
Thinning of the bone (osteoporosis) which could lead to weakness of the bone, bone fractures or delay in healing of fractures
Inflammation of the pancreas which can lead to severe abdominal pain and in some very rare cases can be fatal
Damage to the muscle which can release a protein that can cause severe damage to the kidneys
Inflammation of the intestinal tract which can result in diarrhea and bleeding
This drug can cause severe birth defects in a developing fetus, if you are capable of becoming pregnant or of child bearing age, you must practice 2 forms of reliable birth control, sign the Patient
Slowed growth
Irregular periods
Mild kidney damage which could lead to blood or protein in the urine or renal stones
Extra bone growth along the spine and a tendency for calcium deposits in the tendons and ligaments where they attach to the bone which can lead to pain or stiffness and arthritis of the back and tendonitis
Information/Informed Consent form(s), have regular pregnancy tests, be able to keep appointments and agree to follow the iPLEDGE program steps (a special program required by the manufacturers and approved by the Food and Drug Administration (FDA) which your doctor will explain to you).
Risks and side effects related to melphalan include those which are:
Likely
Less Likely
Rare But serious
- Loss of Appetite
- Nausea and/or vomiting
- Low levels of salt in the blood which may need to be treated (usually associated with high doses)
- Diarrhea
- Inflammation and/or sores in the mouth (and/or throat and/or esophagus, the tube that leads from the mouth to the stomach) that may make swallowing difficult and are painful (painful mouth sores)
- Temporary hair loss
- Fewer white blood cells, red blood cells and platelets in the blood
- a low number of white blood cells can make it easier to get infections
- a low number of red blood cells can make you feel tired and weak
- a low number of platelets causes you to bruise and bleed more easily
- Absence of menstrual cycles (periods) and damage to the ovaries that may decrease the ability to have children in the future
- Absence or decrease in the number of sperm which may be temporary or permanent which may decrease the ability to have children
- Inability to have children (infertility)
- Sweating
- Itching
- Low blood pressure
- Abnormal heart rate (usually associated with high doses)
- Damage to the skin if the medication leaks from the vein
- Increase in the blood of certain enzymes or bilirubin (a substance that comes from the liver breaking down waste products) which could indicate liver irritation or damage
- Severe allergic reaction which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, chills and fever
- Seizures
- Damage to the liver which can lead to inflammation and/or scarring which could lead to a yellow appearing skin, and fluid collection in the abdomen (belly) which makes it look larger
- Severe damage to the bone marrow which could lead to low numbers of white blood cells, red blood cells and platelets and could be permanent
- Sudden damage to the red blood cells (hemolytic anemia) which could cause a rapid decrease in the number of red blood cells such that you would be tired and weak and feel short of breath and may require a blood transfusion
- Inflammation and/or scarring of the lungs that can lead to fluid in the lungs and affect your ability to breath and the levels of oxygen in your blood making you short of breath
- A new cancer or leukemia resulting from this treatment
Risks and side effects related to myeloid growth factors (filgrastim (G-CSF) or pegfilgrastim) include those which are:
Likely
Less Likely
Rare but serious
- Aching or pain in the bones
- Pain, redness, itching, and hardening of the skin and bruising at the site of the injection
- Headache
- Higher than normal levels of liver enzymes in the blood which may indicate liver irritation or damage
- Increase of uric acid in the blood
- A low number of platelets in the blood which may cause you to bruise and bleed more easily
- Low fever
- Enlargement of the spleen (an organ in the abdomen/belly which stores blood cells) which may cause pain in the abdomen or left shoulder
- Higher than normal white blood count
- Skin condition marked by fever and painful skin lesions that appear mainly on the face, neck, back and arms
- Rash or worsening of rash
- Inflammation of blood vessels in the skin leading to a raised purple rash and bruising has been seen mainly in patient who are treated for a long time
- Overall reddening with feelings of warmth
- Allergic reactions which can be life threatening with shortness of breath, low blood pressure, rapid heart rate, hives, itching, and facial swelling.
- Serious allergic reaction which can be life threatening with rapid build-up of fluid under the skin, in the lining of the intestine, and possibly in the throat or swelling of the tongue which could make it difficult to breath
- If you are known to have sickle cell disease, filgrastim or pegfilgrastim may cause a sickle cell crisis.
- Severe damage to the spleen (an organ in the abdomen/belly which stores blood cells) which could lead to pain and loss of blood into the abdomen (belly) and maybe life threatening
- Difficulty breathing and lung damage that may be due to the white blood cells that are stimulated by filgrastim or pegfilgrastim traveling to the lungs when they are inflamed or infected.
- A blood disorder or leukemia that has only been seen in patients with certain immune disorders who are treated for a very long time
Attachment #2
Risks of Radiation Therapy and Peripheral Blood Stem Cell Harvesting and Reinfusion
Radiation Therapy Risks
The risks of radiation therapy depend on the parts of the body being treated. Some possible risks are described below, but you should talk to your child's doctor to see which apply to your child. Radiation therapy can cause nausea, vomiting, diarrhea, red or dry skin, low blood counts, hair loss (permanent or temporary), jaw pain and swelling, temporary weakness or loss of sensation. Some patients have a week or two of low grade fever and sleepiness can occur six to eight weeks after radiation therapy is done. Damage to body organs such as the brain, eyes, heart, lung, liver, and kidneys can occur. Radiation therapy can also cause abnormal bone growth. There is also a small chance that radiation can cause another type of tumor years later.
Peripheral Blood Stem Cell Harvesting and Reinfusion
These procedures are usually safe. Side effects that can occur during PBSC collection include nausea, vomiting, fainting or dizziness, seizures, skin rash, hives, flushing (redness and warmness of the skin, usually the face), blood loss, and infection. Tingling of the lips, muscle cramping and, very rarely, changes in the heart rhythm can occur. These can be prevented or made milder by giving calcium supplements, either by mouth or IV. Very rarely, (less than 1 in 1,000 procedures), clotting may occur in the apheresis machine or in a patient and is potentially life-threatening. To reduce the risk of clotting, you will be given a drug called ACD (acid-citratedextrose). This drug may increase the risk of bleeding and may cause temporary tingling of the lips and limbs, muscle cramping, seizures, or changes in the heart rhythm.
The risks associated with infusing the cells back into your body include dark urine, nausea, vomiting, fever, chills, and high blood pressure. All of these are temporary and go away after the infusion is done. As with any procedure, there may be side effects we do not expect.
Attachment #3
Certificate of Confidentiality Information
The Children's Oncology Group has received a Certificate of Confidentiality from the federal government, which will help us protect the privacy of our research subjects. The Certificate protects against the involuntary release of information about subjects collected during the course of our covered studies. The researchers involved in the studies cannot be forced to disclose the identity or any information collected in the study in any legal proceedings at the federal, state, or local level, regardless of whether they are criminal, administrative, or legislative proceedings. However, the subject or the researcher may choose to voluntarily disclose the protected information under certain circumstances. For example, if the subject or his/her guardian requests the release of information in writing, the Certificate does not protect against that voluntary disclosure. Furthermore, federal agencies may review our records under limited circumstances, such as a DHHS request for information for an audit or program evaluation or an FDA request under the Food, Drug and Cosmetics Act. The Certificate of Confidentiality will not protect against the required reporting by hospital staff of information on suspected child abuse, reportable communicable diseases, and/or possible threat of harm to self or others.
@ - Please note that this test is only recommended to be done on Days 1, 2, 3 and 4
SEE PROTOCOL SECTION 5.0 FOR DOSE MODIFICATIONS. SEE APPENDIX II FOR SUPPORTIVE CARE
NOTE: For eRDEs reporting: use Day 22 (or treatment start date for next cycle if > Day 22) as the end date for the current cycle.
1 Obtain 2x weekly while on G-CSF (recommended)
2 Obtain as scheduled and daily during cisplatin administration (recommended)
3 Obtain serum albumin pre-treatment only and then as clinically indicated.
4 No restriction on method for GFR calculation. If a creatinine clearance is performed at end of induction/prior to consolidation and the result is , the test must be repeated and a GFR performed. The GFR must be obtained by blood sampling method or iothalamate clearance method for all patients with GFR/creatinine clearance
*The conversion of the Lansky to ECOG scales is intended for NCI reporting purposes only.
You will be given a separate consent form for part 2 of the study.
Reported with filgrastim Reported with pegfilgrastim
Reported with filgrastim Reported with pegfilgrastim